Определение ΔH° по зависимости константы равновесия от температуры
1. Цель темы
Понять, как по изменению константы равновесия с температурой можно определить стандартное изменение энтальпии реакции (ΔH°), используя интегрированное уравнение Вант-Гоффа.
2. Основные понятия
Константа равновесия (K)
Отношение произведения равновесных активностей продуктов реакции к произведению активностей реагентов, возведённых в степени их стехиометрических коэффициентов.
Для точного определения ΔH° константа равновесия должна быть выражена через активности. При работе с разбавленными растворами допустимо использовать концентрации, но при высокой ионной силе возможны систематические ошибки.
Стандартная энтальпия реакции (ΔH°)
Тепловой эффект реакции, протекающей при стандартных условиях (давление 1 бар, стандартные состояния реагентов и продуктов).
Температура (T)
Абсолютная температура, выражается в кельвинах (K).
Газовая постоянная (R)
R = 8.314 Дж/(моль·K)
3. Дифференциальное уравнение Вант-Гоффа
Исходным пунктом является термодинамическое соотношение между изменением стандартной энергии Гиббса и константой равновесия:
ΔG° = -RT ln K
Беря производную по температуре и используя уравнение Гиббса-Гельмгольца, получаем:
d(ln K)/dT = ΔH°/(RT²)
Это дифференциальная форма уравнения Вант-Гоффа, которая показывает, как скорость изменения константы равновесия с температурой связана с энтальпией реакции.
4. Интегрированное уравнение Вант-Гоффа
В предположении, что ΔH° не зависит от температуры в исследуемом интервале, уравнение можно проинтегрировать. Результатом интегрирования являются две эквивалентные формы:
Форма линейного уравнения:
ln K = -ΔH°/R × 1/T + ΔS°/R
Форма для двух точек:
ln(K₂/K₁) = -ΔH°/R × (1/T₂ - 1/T₁)
5. Физический смысл и графическое представление
Уравнение ln K = -ΔH°/R × 1/T + const является уравнением прямой линии в координатах ln K от 1/T.
Наклон прямой равен -ΔH°/R
Отрезок, отсекаемый на оси ординат равен ΔS°/R
Таким образом, по экспериментальным данным K(T) можно определить:
ΔH° = -R × (наклон графика)
График зависимости ln K от 1/T
6. Экспериментальное определение ΔH°
Измеряют константы равновесия K при нескольких температурах
Строят график в координатах ln K от 1/T
Проводят наилучшую прямую линию через экспериментальные точки
Рассчитывают наклон этой прямой
Вычисляют энтальпию реакции: ΔH° = -R × наклон
7. Калькулятор ΔH°
Введите значения констант равновесия и соответствующие температуры для расчета ΔH°:
Результат расчета:
8. Характер зависимости и принцип Ле Шателье
Если ΔH° > 0 (реакция эндотермическая) — наклон графика отрицательный, и константа равновесия увеличивается с ростом температуры.
Если ΔH° < 0 (реакция экзотермическая) — наклон графика положительный, и константа равновесия уменьшается с ростом температуры.
Это полностью соответствует принципу Ле Шателье: система, на которую воздействуют, смещает равновесие в направлении, противодействующем этому воздействию. Нагрев (увеличение T) смещает равновесие в сторону поглощения тепла, т.е. в сторону эндотермической реакции.
9. Пример расчета
Задача:
Для реакции N₂O₄ ⇄ 2NO₂ измерены константы равновесия: при T₁ = 298 К, K₁ = 10; при T₂ = 323 К, K₂ = 100. Найдите ΔH°.
Вывод по примеру: Положительное значение энтальпии реакции указывает на то, что реакция диссоциации N₂O₄ является эндотермической.
10. Выводы и ограничения
Интегрированное уравнение Вант-Гоффа — это удобный и наглядный способ экспериментального определения стандартной энтальпии реакции. Оно связывает термодинамические величины (ΔH°, ΔS°) с измеряемыми параметрами (K и T).
Важные ограничения и предупреждения:
Постоянство ΔH°: Уравнение справедливо, только если энтальпия реакции ΔH° постоянна в исследуемом температурном интервале. Если экспериментальные точки на графике ln K от 1/T заметно отклоняются от прямой линии, это указывает на зависимость ΔH° от температуры. Это происходит из-за изменения теплоёмкости системы (ΔCₚ ≠ 0).
Недопустимость экстраполяции: В таких случаях использование интегрированной формы уравнения даёт лишь среднее значение ΔH° в измеренном интервале температур. Экстраполяция за его пределы недопустима.
Термодинамическая константа: Для точного определения ΔH° следует использовать термодинамическую константу равновесия, выраженную через активности.
Метод начальных скоростей: определение порядка реакции по экспериментальным данным
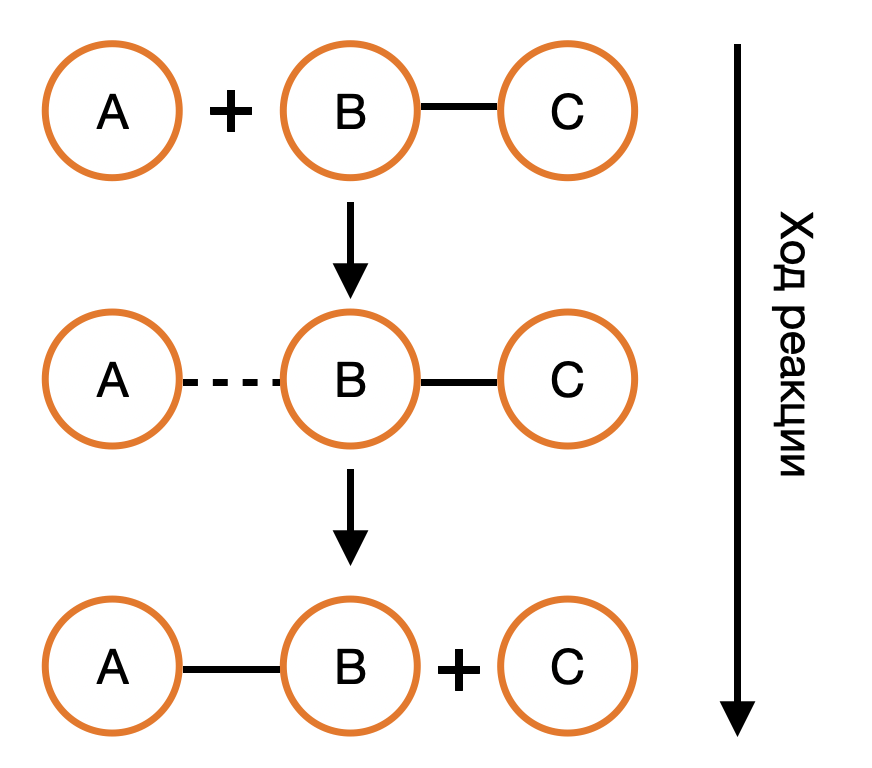
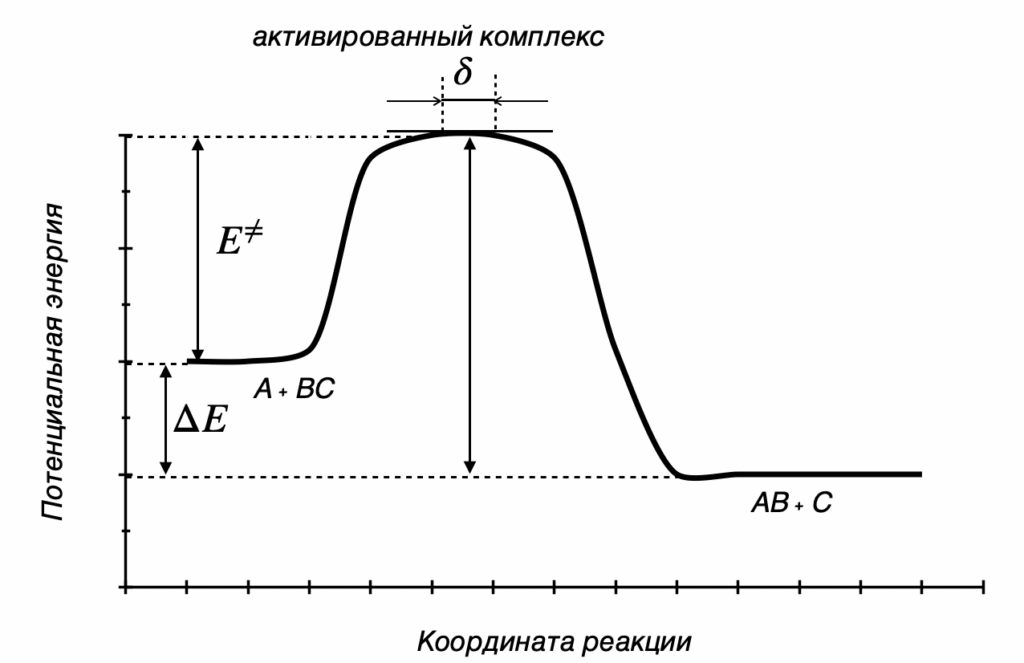
Термодинамика отвечает на вопрос о возможности протекания химического процесса, но не описывает как быстро она проходит. Возможность реакции определяется изменением термодинамических потенциалов — прежде всего, энергии Гиббса (при постоянных температуре и давлении) и энергии Гельмгольца (при постоянной температуре и объёме). Если изменение этих термодинамических потенциалов отрицательно, то процесс термодинамически возможен. Однако на практике далеко не все такие реакции протекают с заметной скоростью: некоторые могут продолжаться годами или вовсе не наблюдаться при невысоких температурах. Это потому, что в термодинамике не учитывается энергия, которая нужна, чтобы реакция началась — так называемый энергетический барьер или энергия активации. Поэтому, чтобы понять, как реально протекает реакция, нужно рассматривать не только её термодинамические свойства, но и кинетику, то есть скорость и условия, при которых она идёт. Центральным понятием химической кинетики является скорость реакции.
Скорость химической реакции — это величина, показывающая, как изменяются концентрации исходных веществ или продукта реакции за единицу времени. скорость (за промежуток времени Δt):
v = ± ΔC / Δt
ΔC — изменение концентрации (моль/л)
Δt — промежуток времени (с, мин)
Знак «+» ставится для продуктов реакции
Знак «–» ставится для исходных веществ
Факторы, влияющие на скорость реакции:
– Концентрация реагентов
–Температура
–Природа реагирующих веществ
–Катализаторы
Однако при анализе реакций возникает практическая задача: определить, как именно концентрации исходных веществ влияют на скорость, то есть установить порядок реакции.
Какие бывают порядки реакций?
1. Нулевой порядок (n = 0)
Кинетическое уравнение: v = k
Единицы измерения k: моль/(л·с)
Скорость не зависит от концентрации реагентов.
форма: [A] = [A]₀ — kt
Примеры: реакции на поверхности катализатора (при полном насыщении поверхности).
Примеры: Реакция щелочного гидролиза сложных эфиров, разложение HI.
4. Дробный порядок
Кинетическое уравнение: например, v = k[A]0,5
Единицы измерения k: л⁰ᐧ⁵/(моль⁰ᐧ⁵·с)
Характерен для сложных реакций (цепных, радикальных).
Пример: Разложение этана (n = 1.5).
5. Общий и частные порядки
Максимальный порядок реакции, который можно наблюдать на практике, равен 3. Реакции более высоких порядков (4, 5 и т.д.) практически неизвестны. Причина кроется в молекулярности — числе частиц, которые должны одновременно столкнуться для протекания реакции. Вероятность одновременного столкновения двух частиц достаточно высока. Подавляющее большинство реакций имеют молекулярность 2 (например, H₂ + I₂ → 2HI). А вот вероятность одновременного столкновения трех частиц в одной точке пространства с правильной ориентацией и достаточной энергией уже очень мала. Такие реакции редки, но известны (например, 2NO + O₂ → 2NO₂). Вероятность одновременного столкновения четырех и более частиц ничтожно мала. Такого события при обычных условиях можно ждать практически бесконечно долго.
Но бывают же реакции 4-го и более высоких порядков?
Если мы экспериментально определяем порядок, например, 4, это не означает, что 4 молекулы столкнулись одновременно. Это означает, что реакция сложная (многостадийная) и ее механизм включает несколько быстрых промежуточных стадий (часто с образованием промежуточных соединений). Суммарный порядок такой сложной реакции является комбинацией порядков отдельных стадий и может быть дробным или целым, больше 3.
Классический пример «реакции 3-го порядка»:
2NO + O₂ → 2NO₂
Молекулярность — понятие, относящееся к элементарной стадии (простой реакции, идущей в одну стадию).
Порядок — понятие, относящееся к реакции в целом (которая может быть сложной и многостадийной).
Для элементарной (простой) реакции порядок совпадает с молекулярностью. Если реакция сложная (состоит из нескольких стадий), ее порядок определяется самой медленной (лимитирующей) стадией и является результатом сложного механизма, поэтому он может не совпадать с молекулярностью и быть дробным.
Чтобы рассчитать порядок реакции в химической кинетике используется
метод начальных скоростей
— экспериментальный подход, позволяющий по данным о скоростях при различных начальных концентрациях реагентов определить индивидуальные и общий порядки реакции, а также рассчитать константу скорости.
Для начала сформулируем Закон Действующих Масс:
Для элементарной реакции скорость пропорциональна произведению концентраций реагентов в степенях, равных их стехиометрическим коэффициентам. Для сложной реакции порядок определяется экспериментально и может не совпадать со стехиометрическими коэффициентами.
Для элементарной реакции: aA + bB → продукты
Скорость реакции описывается уравнением:
v = k × [A]ᵃ × [B]ᵇ
где:
v — скорость химической реакции
k — константа скорости реакции (зависит от температуры и природы реагирующих веществ)
[A], [B] — молярные концентрации реагентов
a, b — стехиометрические коэффициенты в уравнении реакции
показатели степени a, b в уравнении называют порядками реакции по веществам А,В соответственно, а их сумма называется общим или суммарным порядкомреакции. Значит порядок реакции равен сумме показателей степеней для зависимости скорости реакции от концентрации исходных веществ.
Итак, суть метода начальных скоростей заключается в определении порядка реакции путем измерения скорости в самый начальный момент времени при различных начальных концентрациях реагентов. Этот подход позволяет исключить влияние побочных процессов и продуктов реакции, так как измерения проводятся когда концентрации реагентов точно известны и равны начальным, а обратная реакция еще не успевает протекать.
Запишем скорость хим реакции по здм
v = k × [A]ᵃ × [B]ᵇ (1)
k — константа скорости а показатели степени a,b — неизвестные порядки
Идея метода в том что мы измеряем скорость в самый «начальный» момент, когда время стремится к 0.
Запишем уравнение для начальной скорости v₀:
v₀ = k * [A]₀α * [B]₀β (2)
проведем серию опытов, где концентрация вещества В постоянна, а концентрация А меняется
Перепишем уравнение (2) для двух таких опытов:
v₀₁ = k * [A]₀₁α * [B]₀β (3)
v₀₂ = k * [A]₀₂α * [B]₀β (4)
Поделим уравнения (4) на (3) (Сокращаются k и концентрации В)
(v₀₂ / v₀₁) = ([A]₀₂ / [A]₀₁)α (5)
Прологарифмируем 2 части уравнения чтобы найти α
ln(v₀₂ / v₀₁) = α * ln([A]₀₂ / [A]₀₁) (6)
α = ln(v₀₂ / v₀₁) / ln([A]₀₂ / [A]₀₁) (7)
Для большей точности используют не 2 точки а график
Аналогично проведем определение порядка по веществу В
β = ln(v₀₂ / v₀₁) / ln([B]₀₂ / [B]₀₁) (7`)
Теперь можно найти общий порядок реакции, нужно сложить получившиеся частные порядки
n = α + β (8)
Найдем константу скорости k и выразим v₀
k = v₀ / ([A]₀α * [B]₀β (9)
v₀ = k * [A]₀α * [B]₀β (9`)
Прологарифмировав уравнение начальной скорости получим функцию для графика
При постоянном [B]₀ уравнение превращается в:
ln(v₀) = const + α * ln([A]₀) (10)
,где const = ln(k) + β*ln([B]₀)
Пример: Определение порядка реакции окисления йода пероксидом водорода
Реагент A: Иодид-ион (I⁻) (например, из KI — йодида калия)
Реагент B: Пероксид водорода (H₂O₂)
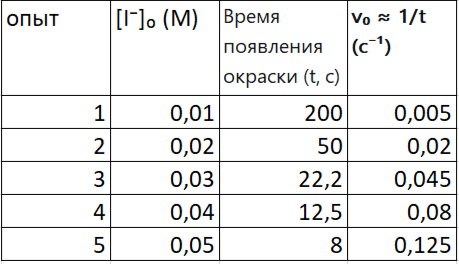
Фиксируем [H₂O₂]₀ = 0.02 M, [H⁺]₀ = 0.01 M (во всех опытах), а меняем — концентрацию [I⁻]₀.
А как измерить скорость этой реакции?
Реакцию можно проводить в присутствии крахмала. Он с продуктом (I₂) дает синее окрашивание. Засекаем время появления синей окраски. В данном эксперименте начальную скорость v₀ можно считать пропорциональной 1/t, где t — время появления окраски. Это справедливо как приближение, только если количество продукта (I₂), необходимое для появления заметной окраски, одинаково во всех опытах. В общем случае скорость определяется как v = Δ[продукт]/Δt.
таблица 1
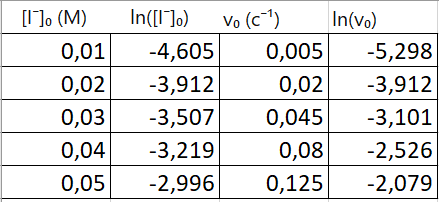
таблица 2
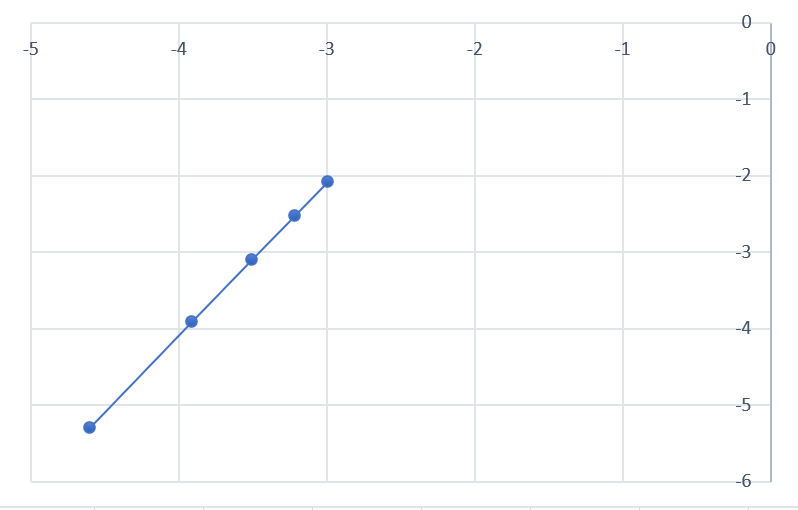
Строим график в координатах ln(v₀) (ось Y) от ln([A]₀) (ось X), у нас должна получиться прямая линия (независимо от порядка реакции) тангенс наклона которой равен частному порядку α
Угловой коэффициент (наклон) этой прямой будет равен ≈ 2.
Вывод: Частный порядок реакции по иодид-иону (I⁻) равен 2. Реакция второго порядка по [I⁻]. (то же можно проделать для В)
Преимущества метода начальных скоростей
Исключает мешающие факторы: Скорость измеряется в начальный момент (t→0), когда:
Нет влияния продуктов реакции и обратной реакции.
Концентрации реагентов точно известны и равны начальным.
Прямое определение частных порядков: Позволяет найти порядок по каждому реагенту в отдельности (α, β), а не только общий порядок (n).
Упрощает математику: Не требует интегрирования сложных кинетических уравнений, в отличие от интегрального метода.
Универсальность: Подходит для изучения сложных реакций (обратимых, параллельных, ступенчатых), где интегральные методы неприменимы.
Выводы:
Метод начальных скоростей является эффективным инструментом для определения частных и общего порядков химической реакции, что было подтверждено на практике.
графики линейны, это подтверждает справедливость степенного закона (закона действующих масс) для данной реакции.
Вычисление рН и рОн буферных систем. Уравнение Гендерсона-Гессельбаха
Каждая из буферных систем характеризуется определенной присущей ей концентрацией ионов [Н+].Эту концентрацию система стремится сохранить на неизменном уровне при добавлении к ней сильной кислоты либо щелочи. pH=-lg[H+]
Буферный раствор-это раствор, который способен поддерживать постоянное значение pH при добавлении небольших количеств кислоты или щелочи, а также при разбавлении.
Буфер характеризуется буферной ёмкостью — количеством сильной кислоты или основания, которые следует прибавить к 1 л буферного раствора, чтобы его pH изменился на единицу. Буферная ёмкость тем выше, чем больше концентрация компонентов раствора. Буферы бывают кислотные и основные, представляют собой смесь кислоты или основания и сопряжённых с ними соли соответствующего остатка.
Вывод уравнения Гендерсона-Гассельбаха
Установим на примере ацетатного буфера факторы, влияющие на величину pH: CH3COO ⇄CH3COO– + H+ CH3COONa→ CH3COO– + Na+ Соль является сильным электролитом, поэтому диссоциирует полностью Реакция диссоциации слабой уксусной кислоты обратима и характеризуется соответствующей Ka.
Ka = [CH₃COO–][H+]/[CH₃COOH]
Т.к. соль CH3COONa диссоциирует нацело,то именно она вносит основной вклад в [CH3COO—], поэтому равновесную концентрацию [CH3COO—] можно представить, как CM(CH₃CONa). Значит [CH3COO—] =CM(CH₃CONa) В правой части уравнения диссоциации уксусной кислоты есть [CH3COO—],основной вклад в которую вносит диссоциация соли,согласно принципу Ле-Шателье равновесие реакции диссоциации уксусной кислоты смещается влево,т.е. в сторону образования ее молекул. Причем диссоциация уксусной кислоты в присутствии собственной соли может быть настолько подавленной, что равновесную концентрацию ее нераспавшихся молекул в растворе можно считать равной концентрации:[СН3СООН]=CM(СН3СООН) Имеем:
Ka = [CH₃COO–][H+]/[CH₃COOH]
[CH3COO—] =CM(CH₃CONa)
[СН3СООН]=CM(СН3СООН) В подстановке получаем: Ka(CH3COOH)=CM(CH₃CONa)[H+]/CM(СН3СООН) Выражаем [H+]: [H+]=Ka(CH3COOH)*CM(СН3СООН)/CM(CH₃CONa) CM(СН3СООН)/CM(CH₃CONa)-данное выражение характеризует буферную ёмкость раствора. Логарифмируя по десятичному логарифму полученное выражение,взятое со знаком (-) получаем: -lg[H+]=-lgKa(CH3OOH)-lgCM(СН3СООН)/CM(CH₃CONa) -lg[H+]=pH -lgKa(CH3OOH)=pKa(CH3COOH) Имеем: pH=pKa(CH3OOH)-lgCM(СН3СООН)/CM(CH₃CONa) Получили выражение для pH буферного раствора односновной кислоты и ее соли. В общем виде: pH=pKa(к-ты)+lg(CM(соли)/CM(к-ты)) В водных ра-х pH и pOH сопряжённые велечины: pH+pOH=14 Получается,что можно написать уравнение для pOH : pOH=14-pKa+lg(CM(к-ты)/CM(соли)) Аналогично можно вывести pOH буферного раствора основания и его сопряженной соли,используя константу диссоциации слабого основания(Kb): pOH=pKb-lg(CM(осн)/CM(соли)) Или pH=14-pKb+lg(CM(осн)/CM(соли))
Анализ уравнения
1)Из приведенных выше уравнений следует, что рН и рОН буферной системы зависит от константы кислотности или основности слабого электролита, входящего в ее состав и от соотношения концентраций компонентов буфера. Значение рК для слабого электролита является величиной постоянной, не зависит от концентрации этого электролита в растворе и приводится в соответствующих справочниках. Зная его можно с помощью уравнения Гендерсона-Гассельбаха рассчитать рН буферного раствора, если известен его количественный состав или, наоборот, определить состав раствора (исходные концентрации его компонентов, который будет обеспечивать заданное значение рН. На практике обычно пользуются готовыми таблицами, в которых указано, в каком соотношении должны быть взяты компоненты для получения буферного раствора с желаемым значением рН.
2)Изменяя концентрацию какого-либо компонента можно сместить значение рН в ту или иную сторону для достижения нужной величины. В буферных системах, используемых на практике, концентрации компонентов не отличаются друг от друга более чем в 10 раз, т.е. их рН не отклоняется больше чем на единицу от величины рK своего слабого электролита. Таким образом, область практических значений рН буферных систем (область буферирования) лежит в интервале pK ± 1. Если концентрации компонентов буферного раствора различаются более чем в 10 раз, то такой раствор обладает слабым буферным действием и может удерживать неизменным содержание ионов (Н+) только при добавлении очень малых иколичеств сильной кислоты либо щелочи. Это делает неудобным его использование в практических целях.
Допустимость применения уравнения
1) если кислота либо основание буферной системы не является достаточно слабым электролитом (например, для кислоты pKa < 3). Тогда нельзя пренебрегать их диссоциацией в присутствии собственной соли. При точном расчёте pHв буфере нужно учитывать коэффициенты активности особенно при I > 0.01 М. Как это сделать можно увидеть в статье
2) если кислота либо основание буферной системы являются, наоборот, слишком слабыми электролитами (например, для кислоты pKa > 11). Тогда нельзя пренебрегать гидролизом их солей.
Примеры задач с использованием уравнения Гендерсона-Гассельбаха
1)Необходимо найти массу навески для приготовления буферного раствора с определенной ph Какую навеску ацетата натрия следует растворить в 1л раствора уксусной кислоты с концентрацией 0,04М, чтобы получить раствор с ph=5 Решение: Распишем уравнение Гендерсона-Гассельбаха для нашей системы(CH3COOH/CH3COONa): pH=рКа+lg(CM(H3CCOONa)/CM(CH3COOH)). При смешении навески CH3COONa,будем считать, что объем раствора не меняется, т.к. вклад навески соли в объем раствора CH3COOH незначительный. Выразим CM(CH3COONa) через массу навески соли CM(CH3COONA)=m(CH3COONA)/(M(CH3COONA)*V(CH3COOH))
CM(CH3COONA)=m(CH3COONa)/82*1 Представляем в уравнение Гендерсона-Гассельбаха с учётом pKa(CH3COOH), ивзятого из справочника: 5=4,76+lg(CM(CH3COONa)/0,04) Получаем: CM(CH3COONA)=m(CH3COONa)/82*1=0,69512М Тогда m(CH3COONA)=CM(CH3COONa)*82*1 m(CH3COONA)=0,69512*82*1=56.99гр
2) Необходимо найти ph буфера,зная концентрации веществ Рассчитать pH ацетатного буферного раствора, приготовленного из 80мл 0,1н раствора CH3COOH и 20мл 0,1н раствора CH3COONA. Решение, посчитаем CM веществ в растворе при смешении. CM‘(CH3COOH)=Cn(CH3COOH)V(CH3COOH)/V(CH3COOH)+V(CH3COONA)
Уравнение Дебая–Хюккеля: расчёт коэффициента активности в разбавленных растворах.
Предпосылки возникновения теории
Поскольку реальные растворы (включая электролиты) не являются идеальными из-за взаимодействий между частицами. Для расчётов было введено понятие коэффициент активности, а состояние в компонента в системе стали описывать химическим потенциалом.
Уравнение для реального раствора:
где,
μk— химический потенциал компонента «k»
μ0 — стандартный химический потенциал компонента раствора вида k
R — универсальная газовая постоянная
T — абсолютная температура
Nk— мольная доля компонента «k»
γ± — средний ионный коэффициент активности
Средний ионный коэффициент активности вводится потому, что измерить коэффициент для отдельного иона невозможно.
Для электролита вида Mν+Aν−
γ+, γ− — коэффициенты активности катиона и аниона
ν+, ν−— стехиометрические коэффициенты катиона и аниона
Запомните: коэффициенты активности зависит от ионной силы, но не зависят от вида иона.
Основные положения теории Дебая-Хюккеля
Цель теории: Рассчитать коэффициент активности ионов.
Уравнение связывает химические потенциалы иона в реальном и идеальном растворе с работой по его переносу:
Δμi — изменение химического потенциала иона
μ1 -химический потенциал в реальном растворе
μ2 -химический потенциал в идеальном растворе
RTlnγi — работа по его переносу 1 моля ионов из реального раствора в идеальный.
ΔGi — изменение энергии Гиббса системы ni— количество вещества компонента i
Допущения теории: 1.Степень диссоциации сильных электролитов равна единице. 2. Ионы- точечные заряды. 3. Учитываем только кулоновские силы и тепловое движение. 4. Диэлектрическая проницаемость раствора постоянна (как у чистого растворителя).
Приближения теории Дебая-Хюккеля
Математическим результатом теории стали уравнения для расчета среднего ионного коэффициента активности, вид которых зависит от диапазона концентраций.
Первое уравнение — позволяющее вычислить средний ионный коэффициент активности
где,
γ± — средний ионный коэффициент активности
h — константа, зависящая от диэлектрической проницаемости раствора (ε) и температуры
z+, z− — заряды катиона и аниона
I — ионная сила раствора
В данное уравнении мы пренебрегаем конечными размерами ионов Оно справедливо при I<0,01 моль/л Запомните: Уравнение предсказывает линейную зависимость lgγ± от √I
Второй уравнение: а)Считаем, что ионы имеют определенный размер. Введём две константы: одну можно рассчитать теоретически, это константа В, которая зависит от температуры и диэлектрической проницаемости среды.
где,
B — константа
ε — диэлектрическая проницаемость растворителя
T — абсолютная температура (в Кельвинах)
5,029*1011 — числовой коэффициент с размерностью, обеспечивающей соответствие единицам измерения
б) Учитываем размер иона:
где,
a — эффективный диаметр иона (расстояние наибольшего сближения электрических центров ионов)
B — константа
A — константа
Параметр a невозможно предсказать теоретически, его определяют экспериментально
3.Уравнение для высоких концентраций:
где C и D — эмпирические параметры, учитывающие дополнительные эффекты при высоких концентрациях.
Ограничения теории Дебая-Хюккеля
1.Применима только к разбавленным растворам. 2.Не учитывает взаимодействия: образование ионных пар, сольватацию, ван-дер-ваальсовы силы. 3.Не предсказывает минимум на кривой зависимости γ± от √I
Пример решения задачи.
Задача: Вычислите средний ионный коэффициент активности (γ±) и величину pH для 0.03 М раствора H₂SO₄ при 298 К.
Решение:
1.Рассчитываем ионную силу (I). H₂SO₄ — сильный электролит, диссоциирует ступенчато, но для расчета ионной силы в первом приближении учитываем полную диссоциацию до 2H⁺ и SO₄²⁻.
Используем второе уравнение, так как ионная сила больше 0.01. Константа A для воды при 25°C равна 0.509. lg γ± = -A * |z₊·z₋| * (√I / (1 + √I)) lg γ± = -0.509 * 2 * (√0.09 / (1 + √0.09)) lg γ± = -1.018 * (0.3 / (1 + 0.3)) = -1.018 * 0.231 ≈ -0.235 γ± = 10-0.235 ≈ 0.582
3. Рассчитываем активность ионов H⁺ и pH.
В данном случае для расчета pH нам нужна активность ионов H⁺ (aₕ₊). aₕ₊ = [H⁺] * γₕ₊ Для бинарного электролита типа 1-2: γ±3 = γₕ₊2 * γₛₒ₄2-. В первом приближении для расчета концентрационной зависимости принимают, что γₕ₊ ≈ γ±. aₕ₊ ≈ [H⁺] * γ± = 0.06 * 0.582 ≈ 0.03492
Определение концентрации ионов с помощью ионселективных электродов (ИСЭ). Уравнение Нернста для ионометрии
Цель урока: понять принцип работы ионселективных электродов и научиться применять уравнение Нернста для количественного определения концентрации ионов в растворе
Введение в ионометрию
Ионометрия – это раздел аналитической химии, занимающийся определением концентрации (активности) ионов в растворе с помощью ионселективных электродов (ИСЭ).Ионселективный электрод (ИСЭ) – это электрохимический датчик, потенциал которого зависит от концентрации (точнее, активности) конкретного иона в растворе. Его ключевая особенность – селективность (избирательность) по отношению к определенному иону (например, H⁺, Na⁺, Ca²⁺, F⁻, NO₃⁻).
Устройство и принцип работы ИСЭ
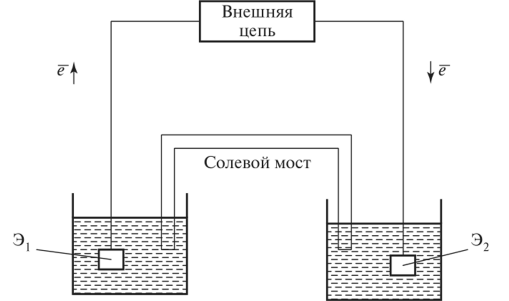
Стандартная электрохимическая ячейка для измерений с ИСЭ содержит 2 электрода:
-индикаторный — электрод,потенциал которого прямо или косвенно зависит от концентрации определяемого вещества
Потенциал ИСЭ возникает на границе раздела фаз между мембраной электрода и анализируемым раствором.Мембрана обладает селективной проницаемостью. Процесс установления потенциала связан с обменом этими ионами между мембраной и раствором
— Два сосуда с растворами, в которых помещены электроды Э1 и Э2.
— Электроды контактируют с растворами и участвуют в электрохимической реакции.
— Между двумя сосудами расположен солевой мост, служащий для ионной проводимости и предотвращения смешивания растворов.
-Внешняя цепь обеспечивает прохождение электронов: электроны движутся от одного электрода к другому (обозначено стрелками e−→).
— Электроды Э1 и Э2 погружены в растворы и формируют полуэлементы, между которыми возникает электрохимический потенциал.
В классической схеме используется два электрода, но в современных датчиках часто применяют комбинированные электроды.Большинство комбинированных ИСЭ (pH-электроды) имеют встроенный электрод сравнения и внутренний раствор, поэтому не требуют двух сосудов и солевого моста.
Уравнение Нернста для ионселективного электрода
Фундаментальная зависимость, описывающая работу ИСЭ, – это уравнение Нернста. Для электрода, селективного к иону с зарядом z , оно имеет вид:
Важное замечание: ИСЭ чувствителен именно к активности иона, а не к его концентрации (C). Активность связана с концентрацией соотношением a = γ * C, где γ – коэффициент активности (γ → 1 в очень разбавленных растворах). На практике в аналитических целях часто проводят измерения в условиях, когда γ ≈ const, и тогда уравнение используют для нахождения концентрации.Постоянство γ обеспечивается добавлением фонового электролита (ионной силы регулятора — ISA, TISAB и т.п.). паапра
Упрощенный вид уравнения Нернста:
При T=298 K (25°C) и замене натурального логарифма на десятичный, уравнение принимает более удобный для расчетов вид:
Знак перед логарифмическим членом:
Для катионов (z > 0 например, H⁺, Na⁺, K⁺):
Для анионов (z <0 , например, F⁻, Cl⁻, NO₃⁻):
Пример расчёта
Пусть электрод чувствителен к ионам калия (K⁺, z = 1).
Практическое применение: калибровочный график
Прямое использование уравнения Нернста затруднено из-за необходимости знать точное значение E⁰ . Поэтому на практике используют метод калибровочного графика.
Порядок действий:
1.Готовят серию стандартных растворов с известной концентрацией определяемого иона.
2.Измеряют потенциал ИСЭ в каждом из этих растворов.
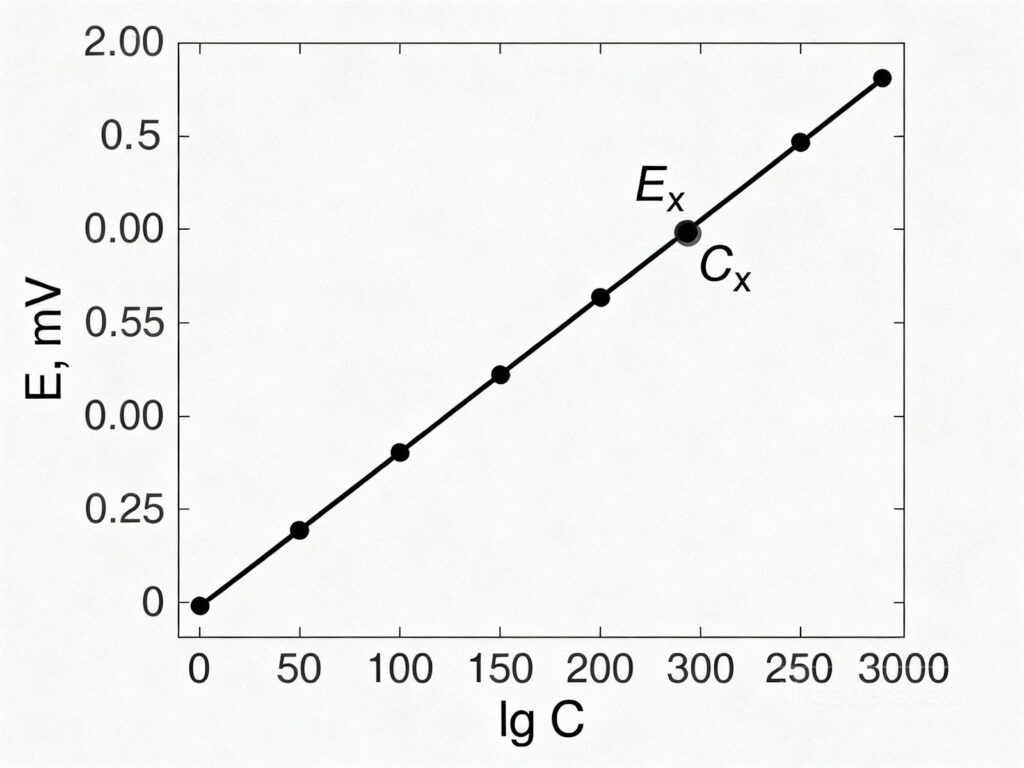
3.Строят график в координатах E = f(lg C).
4.Измеряют потенциал в анализируемом растворе с неизвестной концентрацией Cx
5.По графику находят значение lg Cx , а затем и саму концентрацию Cx .
Рис. Пример калибровочного графика для определения концентрации иона. E = f(lg C).
По измеренному значению Ex находят соответствующее ему значение lg Cx.
По графику Ex соответствует lg Cx=-3, то концентрация неизвестного раствора будет
График представляет собой прямую линию, угол накола которой определяется нернстовской наклоном. Отклонение от теоретического наклона может указывать на неидеальность электрода или мешающее влияние других ионов.
Селективность ИСЭ и мешающие ионы
Ни один ИСЭ не является абсолютно селективным. Потенциал может зависеть от присутствия в растворе других ионов, похожих по свойствам на определяемый. Для учета этого влияния используют модифицированное ур.Нернста
Где:
заряды основного и мешающего ионов —
активность основного иона А и мешающего иона В —
потенциометрический коэффициент селективности (характеризует влияние мешающего иона)-
Этот коэффициент показывает, насколько сильно электрод реагирует на мешающий ион по сравнению с главным.
• Если<< 1 → электрод высокоселективен, мешающий ион почти не влияет.
• Если ≈ 1 → электрод реагирует на оба иона одинаково.
• Если > 1 → мешающий ион оказывает даже большее влияние, чем основной (плохая селективность).
Основные типы ионселективных электродов
Стеклянные электроды: Селективны к ионам H⁺ (pH-метр), Na⁺, K⁺ и др. Действие основано на ионообменных свойствах специального стекла.
Электроды с твердыми мембранами: Мембрана из одного или нескольких кристаллов (например, LaF₃ для F⁻, Ag₂S для S²⁻ и Ag⁺).
Электроды с жидкими мембранами: Мембрана содержит органический раствор селективного ионофора (носителя) в неполярном растворителе. Используются для определения Ca²⁺, NO₃⁻ и др.
Ферментные электроды: Комбинированные датчики, где селективность обеспечивается ферментом, иммобилизованным на поверхности мембраны.
Достоинства и недостатки ионометрии
+
—
Высокая скорость анализа
Влияние мешающих ионов (недостаточная селективность)
Возможность анализа мутных и окрашенных растворов без пробоподготовки
Требуется калибровка
Неразрушающий контроль
Чувствительность к температуре и pH
Широкий диапазон определяемых концентраций (до 5-6 порядков)
Относительно невысокая точность (~2-5%)
Выводы
— Ионселективные электроды – мощный инструмент для прямого и быстрого определения ионного состава растворов.
— Работа ИСЭ описывается уравнением Нернста, связывающим потенциал электрода с логарифмом активности (концентрации) определяемого иона.
— На практике для количественного анализа используют метод калибровочного графика в координатах E от lg C
“Использование уравнения Нернста в окислительно-восстановительном титровании ( пример: перманганатометрия)”
Уравнение Нернста.
Уравнение Нернста описывает зависимость электродного потенциала (E) окислительно-восстановительной пары от концентраций (активностей) её окисленной и восстановленной форм, также важно учитывать ионную силу раствора.
E = E° + (RT / nF) ln([окисленная форма] / [восстановленная форма])
При 25°C: E = E° + (0,0591 / n) log([окисленная форма] / [восстановленная форма])
где E — электродный потенциал; E° — стандартный потенциал; n — число электронов; R = 8,314 Дж/(моль·K); F = 96485 Кл/моль; T — температура (K).
Уравнение Нернста в окислительно-восстановительном титровании используется для расчета потенциала на разных стадиях титрования. Это необходимо для построения кривой титрования и точного определения точки эквивалентности, что обеспечивает высокую точность и надёжность анализа.
В таких титрованиях взаимодействуют окислитель и восстановитель, и в ходе реакции изменяются их концентрации. Поскольку потенциал каждой полуреакции зависит от состава раствора, потенциал системы (E) тоже изменяется.
Окислительно-восстановительное титрование
В основе метода лежат реакции, связанные с перераспределением электронов между участниками. Эти реакции более сложные, так как часто образуются нестойкие промежуточные соединения, которые могут реагировать с другими веществами, находящимися в растворе. Возникают побочные процессы, которые приводят к ошибкам. Скорости этих реакций более медленные, чем кислотно-основные.
Один из методов такого титрования- метод перманганатометрии.
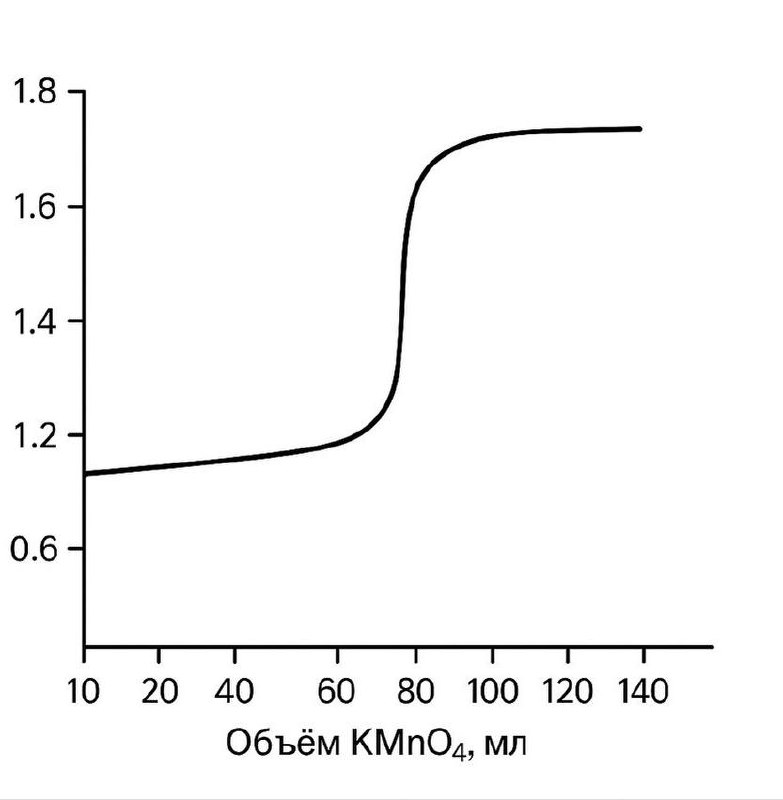
Рабочим раствором в этом методе является перманганат калия. Его окислительные свойства зависит от среды, в которой протекает реакция. Титрование перманганатом калия проводят в кислой среде, так как в этом случае образуются бесцветные ионы Mn2+, тогда как при окислении в щелочной или нейтральной среде выпадает тёмно бурый осадок MnO2 ,что затрудняет фиксирование точки эквивалентности.
При окислительно-восстановительном титровании концентрации реагирующих веществ все время меняются, следовательно, изменяется величина потенциала раствора и кривая титрование строится на основе расчёта электродного потенциала (E0) по уравнению Нернста для различных моментов титрования
aA ок + bB вос = аА вос + bB ок
В любой момент титрования раствор всегда содержит две окислительно-восстановительные формы, следовательно, для вычисления электродного потенциала имеем два уравнения
E = E0 + 0,059\b * lg [Аок]\[Авос] (1)
E=E0 + 0,059\a * lg [Bок]\[Bвос] (2)
Так как потенциал в растворе удовлетворяет обоим уравнением, то для расчёта можно использовать любое, исходя из удобства вычисления концентрации.
До точки эквивалентности пока не оттитровано все вещество B, величину Е определяют по уравнению два.
После точки эквивалентности легче определить концентрацию вещества А и поэтому Е вычисляют по уравнению один.
До начала титрования в растворе присутствует только восстановитель (Fe²⁺), и его окисленная форма (Fe³⁺) отсутствует.
Потенциал формально не определён, но при построении кривой принимают, что [Fe³⁺] ≈ 10⁻⁶–10⁻⁸ М (из-за следов окисления воздухом), и рассчитывают условный стартовый потенциал. Поскольку в чистом растворе Fe²⁺ окисленная форма отсутствует, на практике для построения кривой принимают [Fe³⁺] ≈ 10⁻⁶ M
Точка Эквивалентности. Для вычисления Е в точке эквивалентности в приведённых выражениях один и два уравниваем коэффициенты при числах у логарифмов, затем почленно складываем уравнения
аЕ= aE02 + 0,059lg [Вок]\[Ввос]
bE= bE(0)1 + 0,059lg [Aок]\[Авос]
(а+b) E= aE02 +bЕ01 + 0,059 lg [Вок][Аок]\[Ввос][Авос]
Но в точке эквивалентности на каждый b ионов вещества В ок приходится а ионов А вос, откуда [Вок][Аок]\[Ввост][Авост]=1
Следовательно потенциал в точке эквивалентности: E=bE01 + аЕ02\ а+b
В любой момент титрования потенциалы систем сравниваются: E₁ = E₂.
3.В точке эквивалентности — В точке эквивалентности весь Fe²⁺ окислен до Fe³⁺, и весь MnO₄⁻ восстановлен до Mn²⁺. Потенциал определяется средним значением стандартных потенциалов:
Eэкв = (E°(MnO₄⁻/Mn²⁺) + E°(Fe³⁺/Fe²⁺)) / 2 = (1,51 + 0,77) / 2 = 1,14 В 4. . После точки эквивалентности — избыток перманганата, потенциал определяется парой MnO₄⁻/Mn²⁺.
До эквивалентной точки потенциал определяется парой Fe³⁺/Fe²⁺, после — MnO₄⁻/Mn²⁺. Из-за резкой смены преобладающей пары наблюдается скачок потенциала, что позволяет точно определить конец титрования по изменению окраски (MnO₄⁻ — фиолетовый).
Пара
Полуреакция
E°, В
Уравнение Нернста
MnO₄⁻/Mn²⁺
MnO₄⁻ + 8H⁺ + 5e⁻ → Mn²⁺ + 4H₂O
1,51
E = 1,51 + (0,0591/5) log(([MnO₄⁻]·[H⁺]⁸) / [Mn²⁺])
Fe³⁺/Fe²⁺
Fe³⁺ + e⁻ ↔ Fe²⁺
0,77
E = 0,77 + 0,0591 log([Fe³⁺]/[Fe²⁺])
Итог:
• Уравнение Нернста позволяет количественно описать изменение потенциала в ходе окислительно-восстановительного титрования. • В перманганатометрии потенциал системы определяется соотношением концентраций ионов Fe²⁺/Fe³⁺ и MnO₄⁻/Mn²⁺. • Резкое изменение потенциала указывает на достижение точки эквивалентности. • Метод можно использовать как потенциометрический, так и визуальный (по окраске перманганата).
При построении кривой титрования наблюдается резкий скачок потенциала вблизи точки эквивалентности. Этот скачок используется для потенциометрического определения конца титрования. В перманганатометрии обычно не требуется внешний индикатор, так как избыток KMnO₄ придаёт раствору фиолетовую окраску.
Применение закона Бугера–Ламберта–Бера: расчёт концентрации по оптической плотности
Закон Бугера–Ламберта–Бера (основной закон светопоглощения) применяется для расчета концентрации веществ в растворах на основе измерений их оптической плотности.
Для точного измерения количества вещества с помощью светопоглощения, переводят вещество в форму, способную поглощать свет. Далее измеряют, насколько свет становится слабее, проходя через это вещество на заданное расстояние.Закон Бугера-Ламберта-Бера, который лежит в основе большинства фотометрических методов исследования анализа выражается: I=I0e−^(εcl), где
I — интенсивность светового потока, прошедшего через раствор; I0 — интенсивность светового потока, падающего на кювету определенной толщины; ε — молярный коэффициент поглощения; l — толщина поглощающего слоя; с — концентрация поглощающего слоя;
Молярный коэффициент поглощения (ε) представляет собой фундаментальную константу, которая численно выражает эффективность поглощения светового излучения одним молем анализируемого соединения при строго заданной длине волны.Эта величина ε уникальна для каждого химического вещества и не является постоянной. Её значение существенно варьируется в зависимости от специфической длины волны падающего света, поскольку различные вещества демонстрируют избирательное поглощение излучения в разных участках спектра. Кроме того, на величину молярного коэффициента поглощения оказывают влияние такие факторы, как химический состав растворителя и температурный режим. Важное следствие этого параметра заключается в следующем: чем выше значение ε, тем интенсивнее световой поток будет поглощаться раствором с идентичной концентрацией вещества, при условии сохранения всех прочих экспериментальных параметров (например, одинаковой длины оптического пути). Чаще всего измеряется в л · моль^(-1)· см^(-1) (литры на моль на сантиметр).
Также этот закон можно представить в виде логарифмической формы:
A=ε⋅c⋅l , где
ε — молярный коэффициент поглощения; А — оптическая плотность; с — концентрация поглощающего слоя;
Оптическая плотность, обозначаемая символом А, представляет собой количественную характеристику способности вещества абсорбировать световое излучение, тем самым затрудняя его прохождение. Эта величина является безразмерной. Прямая зависимость такова: чем интенсивнее образец поглощает свет, тем выше численное значение оптической плотности А.
Измерение оптической плотности осуществляется посредством специализированных устройств, известных как спектрофотометры. Сам по себе спектрофотометр — это аналитический инструмент, предназначенный для количественного определения способности исследуемого материала поглощать или пропускать электромагнитное излучение на конкретной длине волны. Конструктивно спектрофотометр объединяет несколько ключевых компонентов, функционирующих вместе для детального анализа светового потока: это источник излучения, монохроматор, камера для кюветы, детектор и блок обработки данных.
● Источник света продуцирует электромагнитное излучение различного спектрального состава (различных длин волн), включая невидимые диапазоны. Его основная функция заключается в освещении исследуемого образца. ● Монохроматор отвечает за то, чтобы к образцу поступал свет строго одной, узкоопределенной длины волны, обеспечивая его спектральную чистоту. ● Кюветное отделение представляет собой специальную камеру, предназначенную для размещения прозрачного контейнера (кюветы) с исследуемой пробой. В этом отсеке световой пучок пронизывает образец, где происходит частичная абсорбция излучения. ● Детектор фиксирует и измеряет интенсивность светового потока, достигшего его после прохождения через образец, тем самым определяя долю излучения, не поглощенного веществом. Большинство спектрофотометров демонстрируют наибольшую точность измерений оптической плотности в диапазоне примерно от 0.2 до 0.8 (или иногда до 1.0). За пределами этого диапазона относительная погрешность измерения оптической плотности значительно возрастает.
Формулировка закона: уменьшение интенсивности света при его прохождении через раствор прямо пропорционален концентрации поглощающего вещества и толщине его слоя
Для количественного определения концентрации вещества в растворе
методом светопоглощения, основанном на законе Бугера-Ламберта-Бера, существуют три основных подхода:
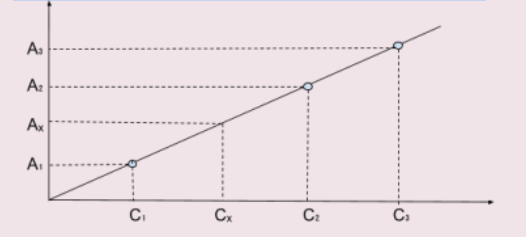
Для определения концентрации вещества при помощи градуировочной кривой готовят серию стандартных окрашенных растворов, концентрация которых охватывает область возможных изменений концентрации исследуемого раствора. Затем измеряют величина оптической плотности А и строят график зависимости оптической плотности от концентрации растворенного вещества в координатах А=f(С). Для построения градуировочной кривой берут 5-8 растворов, отличающихся по концентрации не менее, чем на 30%. Построенную кривую называют градуировочным графиком.
Для построения градуировочной кривой берут 5-8 растворов, отличающихся по концентрации не менее, чем на 30%. Концентрации стандартных растворов выбирают так, чтобы оптическая плотность исследуемого раствора находилась примерно в середине градуировочной кривой. Определив оптическую плотность раствора Ax, находят её значение на оси ординат, а затем соответствующее значение искомой концентрации Cx на оси абсцисс. Этот метод применяют при многократном фотометрировании однотипных по химическому составу растворов при выполнении серийных фотометрических анализов.
Метод дает хорошие результаты при соблюдении закона Бугера-Ламберта-Бера. Важным его отличием от других методов является то, что позволяет определять концентрацию окрашенных веществ даже в тех случаях, когда основной закон светопоглощения не соблюдается. В этом случае для построения градуировочной кривой готовят значительно большее количество стандартных растворов, отличающихся друг от друга не более, чем на 10%.
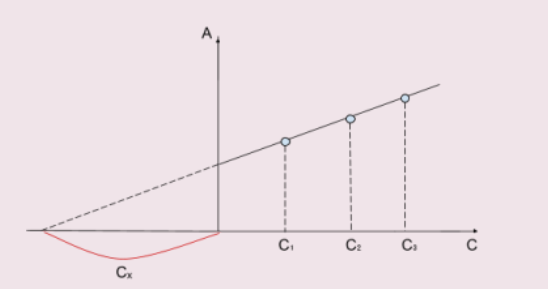
Метод добавок
Определение концентрации раствора этим методом основано на сравнении оптической плотности исследуемого раствора и того же раствора с добавкой известного количества определяемого вещества. Метод добавок обычно применяют либо для упрощения работы, либо для устранения мешающего влияния посторонних примесей. Этом метод позволяет создать одинаковые условия для анализа исследуемого и стандартного (с добавкой) окрашенных растворов. Поэтому его целесообразно применять для определения малых количеств определяемого элемента в присутствии больших количеств посторонних
веществ. Этот метод требует обязательного соблюдения основного закона светопоглощения. Метод добавок, в свою очередь, может быть произведён расчётным путём и графическим.
Расчетный метод
Для начала измеряют оптическую плотность анализируемого раствора, содержащего определённый компонент с неизвестное концентрацией Cx. Затем в анализируемый раствор добавляют известное количество определяемого компонента Сс.т., и вновь измеряют оптическую плотность Ax+с.т. Оптическая плотность Ах анализируемого раствора: (1)Ax=ε⋅l⋅Cx А оптическая плотность анализируемого раствора с добавкой стандартного раствора: (2)Ax+с.т.=ε⋅l⋅(Cx+Сс.т.) Сравним выражения (1) и (2) получим: (3)Cx = Ax⋅Сс.т. /( Ax+с.т — Ax)
Графический метод
Этот способ определения неизвестной концентрации вещества заключается в построении графика, где по оси абсцисс — содержание определяемого компонента, а по оси ординат — оптическая плотность раствора.
Метод сравнения Метод сравнения в аналитике отличается тем, что поглощение образца оценивается относительно эталонного раствора того же вещества с известной, близкой к образцу концентрацией, а не чистого растворителя. Измеряемая в этом случае оптическая плотность, обозначаемая A’, является относительной. Она выражает разность между истинными оптическими плотностями исследуемого и эталонного растворов:
Закон Бугера–Ламберта–Бера строго справедлив только для разбавленных растворов и в определенных условиях:
постоянство состава и неизменность поглощающих частиц в растворе;
Это означает, что вещество, концентрацию которого мы измеряем, должно оставаться в растворе в той же химической форме, что и момент приготовления стандартных растворов
исследуемые молекулы должны быть диспергированы до молекулярного уровня, они не должны рассеивать свет и взаимодействовать друг с другом;
Для точного измерения оптической плотности критично, чтобы исследуемый компонент был в истинном растворе, то есть равномерно распределен на молекулярном или ионном уровне в растворителе. Если же компонент существует в форме нерастворимых микрочастиц (например, в суспензии или коллоидном состоянии), он будет не только поглощать, но и активно рассеивать световой поток. Такое рассеяние приводит к тому, что часть излучения не достигает детектирующего элемента, что в свою очередь приводит к искажению результатов и завышению показаний оптической плотности
монохроматичность и параллельность проходящего через раствор лучистого потока небольшой интенсивности; постоянство температуры. Свет должен состоять из излучения одной, очень узкой длины волны. Если свет полихроматический (много длин волн), а молярный коэффициент поглощения (ε) вещества меняется в зависимости от длины волны (что почти всегда так), то среднее поглощение будет зависеть не только от концентрации, но и от спектрального состава света, нарушая закон.
Когда не соблюдаются условия, при которых Закон Бугера–Ламберта–Бера остается действительным, что часто обусловлено погрешностями в аналитическом процессе, фиксируются расхождения с его предсказаниями. Эти расхождения могут проявляться в двух формах: как увеличение, так и уменьшение ожидаемых значений.
В случае отрицательных расхождений наблюдаемая оптическая плотность оказывается ниже теоретически прогнозируемой Законом Бугера–Ламберта–Бера для заданной концентрации. Визуально это выражается в том, что градуировочная кривая отклоняется вниз при возрастающих концентрациях.
Напротив, положительные расхождения характеризуются тем, что полученная оптическая плотность превышает расчетное значение, которое должно соответствовать текущей концентрации согласно Закону Бугера–Ламберта–Бера. На графике это выглядит как изгиб градуировочной кривой вверх по мере увеличения концентрации
Законы Фарадея: расчет массы вещества при электролизе
Электролиз — это физико-химический процесс разложения вещества (электролита) на составные части при прохождении через его расплав или раствор постоянного электрического тока. Проще говоря, с помощью электричества мы заставляем вещество (которое в обычных условиях не разлагается) распасться на более простые элементы.
Пример: Электролиз раствора CuSO₄ с медными электродами
Установка:
Раствор: Водный раствор CuSO₄. Он содержит ионы: Cu²⁺, SO₄²⁻, H⁺ (от воды), OH⁻ (от воды).
Анод (+): Пластина из неочищенной меди.
Катод (-): Пластина из чистой меди.
Источник тока: Постоянный ток (например, батарея).
Процесс электролиза (шаги):
А) Диссоциация (в растворе): Молекулы соли и воды распадаются на ионы: CuSO₄ → Cu²⁺ + SO₄²⁻ H₂O ⇄ H⁺ + OH⁻
Б) На электродах:
На катоде (-): Катод притягивает положительно заряженные ионы (катионы). У нас есть Cu²⁺ и H⁺. Восстановление: Ион меди Cu²⁺ принимает два электрона и восстанавливается до атома металлической меди. Cu²⁺ + 2ē → Cu⁰ Атомы меди осаждаются на катоде в виде тонкого розово-красного слоя. Катод становится толще и тяжелее.
На аноде (+): Анод притягивает отрицательно заряженные ионы (анионы). У нас есть SO₄²⁻ и OH⁻. Окисление: Однако, анод сделан из меди, и он сам начинает окисляться, теряя электроны, легче, чем анионы. Cu⁰ — 2ē → Cu²⁺ Атомы меди из анода переходят в раствор в виде ионов Cu²⁺. Анод постепенно растворяется и становится тоньше.
Что мы видим: Концентрация сульфата меди (CuSO₄) в растворе не меняется, так как количество ионов Cu²⁺ в растворе остается постоянным (сколько ушло на катод, столько же и пришло с анода).
Суть процесса
В процессе электролиза всегда происходит преобразование электрической энергии в химическую.
Где проводится?
В специальном приборе — электролизере.
Что внутри?
Электролиты: Вещество, которое проводят ток (раствор или расплав соли, щелочи, кислоты).
Электроды: Два проводника, опущенные в электролит.
Анод (+): Положительно заряженный электрод. К нему притягиваются анионы (отрицательно заряженные ионы).
Катод (-): Отрицательно заряженный электрод. К нему притягиваются катионы (положительно заряженные ионы)
Количество электричества (Заряд, q): Измеряется в кулонах (Кл). Для постоянного тока рассчитывается по формуле:
q = I * t , где:
I
сила тока в амперах (А)
t
время в секундах (с)
Законы Фарадея устанавливают количественную связь между количеством электричества, прошедшего через электролит, и массой вещества, выделившегося на электроде.
Первый закон
Масса вещества, выделившегося на электроде, прямо пропорциональна количеству прошедшего через электролит электричества.
m = k * q ,
где:
m
масса выделившегося вещества, [г]
q
количество электричества, [Кл]
k
коэффициент пропорциональности, называемый электрохимическим эквивалентом
Электрохимический эквивалент (k) — это масса вещества, выделяющаяся при прохождении 1 кулона электричества. Это специфическая для каждого вещества постоянная. ВЫВЕСТИ К. РАЗМЕРНОСТЬ
Если пропускать через раствор медного купороса электрический ток в течение определённого количества времени, то на катоде выделяется небольшое количество меди. Однако если пустить ток большей силы, за такое же количество времени на катоде образуется большее количество меди. При увеличении времени и одинаковой силе тока также увеличивается количество меди.
Второй закон
При постоянном количестве прошедшего электричества массы различных веществ, выделяющихся на электродах, пропорциональны их химическим эквивалентам.
Химический эквивалент — это отношение молярной массы вещества (M) к его числу электронов в электродной реакции в данной redox-реакции (z).
Второй закон устанавливает связь между электрохимическим и химическим эквивалентами:
k = M / (z * F) , где:
M
молярная масса вещества, [г/моль
I
сила тока, [А]
k
коэффициент пропорциональности, называемый электрохимическим эквивалентом
z
число электронов, участвующих в электродном процессе
Объединенный закон Фарадея и число Фарадея
Объединив оба закона, можно вывести универсальную формулу для расчета массы вещества при электролизе.
m = (M * I * t) / (z * F) , где:
m
масса выделившегося вещества, [г]
M
молярная масса вещества, [г/моль
I
сила тока, [А]
t
время проведения электролиза, [с]
z
число электронов, участвующих в электродном процессе
F
постоянная Фарадея
Постоянная Фарадея (F) — это количество электричества, необходимое для выделения одного моля эквивалента любого вещества.
F ≈ 96 500 Кл/моль
Это фундаментальная физическая константа, равная заряду одного моля электронов.
Законы Фарадея идеально работают, когда процесс протекает с большой скоростью. Но что, если мы работаем в условиях, близких к равновесию? Здесь нужно использовать уравнение Нернста.
Чаще всего на практике количество электричества Q известно не напрямую, а через силу тока I и время t. Вспомним, что Q = I * t.
Тогда основная рабочая формула принимает вид:
m = (I * t * M) / (F * z)
Шаг 1: Записать полуреакцию на электроде
Определите, какой процесс происходит на интересующем вас электроде (окисление или восстановление), и найдите z – количество электронов, участвующих в реакции для одного иона.
Шаг 2: Определить все известные величины
Выпишите:
I – сила тока (в Амперах, А)
t – время (в секундах, с)
M – молярная масса (в г/моль)
z – заряд иона (безразмерная величина)
F – постоянная Фарадея (96 500 Кл/моль)
Шаг 3: Подставить значения в формулу и произвести расчёт
Пример расчета
Электролиз раствора нитрата серебра (AgNO₃)
Задача: Через раствор нитрата серебра в течение 20 минут пропускали ток силой 2 А. Какая масса серебра выделилась на катоде?
Решение:
Записываем катодную реакцию:
На катоде восстанавливаются катионы серебра:
Ag⁺ + 1ē → Ag⁰
Отсюда видно, что для восстановления 1 моля серебра требуется 1 моль электронов. Значит, z = 1.
Определяем известные величины:
I = 2 А
t = 20 мин = 20 * 60 = 1200 с
M = 108 г/моль (молярная масса серебра)
z = 1
F = 96500 Кл/моль
Подставляем в формулу:
m = (I * t * M) / (F * z)
m = (2 А * 1200 с * 108 г/моль) / (96500 Кл/моль * 1)
Рассчитываем:
m = 259200 / 96500 ≈ 2.69 г
Электролиз раствора сульфата меди (CuSO₄)
Задача: Какую массу меди можно получить при электролизе раствора CuSO₄ за 1 час при силе тока 5 А?
Решение:
Записываем катодную реакцию:
На катоде восстанавливаются катионы меди:
Cu²⁺ + 2ē → Cu⁰
Для восстановления 1 моля меди требуется 2 моля электронов. Значит, z = 2.
Определяем известные величины:
I = 5 А
t = 1 час = 3600 с
M = 64 г/моль (молярная масса меди)
z = 2
F = 96500 Кл/моль
Подставляем в формулу:
m = (5 А * 3600 с * 64 г/моль) / (96500 Кл/моль * 2)
m = (1143000) / (193000) ≈ 5.92 г
3) Рассчитайте массу меди, выделившейся на катоде за 30 минут при силе тока 5 А. Электролизу подвергался раствор сульфата меди (CuSO₄).
Запишем объединенный закон Фарадея: m = (M * I * t) / (z * F)
Определим параметры для меди (Cu):
· M = 64 г/моль
· z = 2 (так как ион Cu²⁺)
· I = 5 А
· t = 30 мин * 60 = 1800 с
· F = 96500 Кл/моль
Подставим значения в формулу:
m = (64 г/моль * 5 А * 1800 с) / (2 * 96500 Кл/моль)
Любая химическая реакция сопровождается энергетическими изменениями. Чтобы предсказать, пойдёт ли реакция при данных условиях, и рассчитать тепловой эффект, мы используем термодинамические функции: энтальпию (H), энтропию (S) и энергию Гиббса (G). Критерием самопроизвольного протекания процесса при постоянных температуре и давлении является уменьшение энергии Гиббса (ΔG < 0).
Стандартные состояния и справочные данные
Все расчёты начинаются с использования стандартных термодинамических величин, которые приведены для температуры 298,15 К (25 °C) и давления 100 кПа.
В справочниках вы найдёте для каждого вещества:
ΔH°f,298 – стандартная энтальпия образования (кДж/моль). Энтальпия изменения при образовании 1 моля вещества из простых веществ в их стандартных состояниях.
S°298 – стандартная энтропия (Дж/(моль·К)). Мера неупорядоченности системы.
ΔG°f,298 – стандартная энергия Гиббса образования (кДж/моль).
Важно: Для простых веществ в их наиболее устойчивой аллотропной форме (O2(г), C(графит), Fe(тв) и т.д.) ΔH°f,298 и ΔG°f,298 принимаются равными нулю.
Расчёт стандартных изменений при 298 К
Для реакции вида: aA + bB → cC + dD
1. Стандартное изменение энтальпии реакции (ΔH°р-ции):
где n – стехиометрические коэффициенты продуктов (пр) и исходных веществ (исх).
Способ 2 (через энтальпию и энтропию, фундаментальное уравнение):
ΔG°р-ции = ΔH°р-ции — T·ΔS°р-ции При T = 298 K: ΔG°298 = ΔH°р-ции — 298·ΔS°р-ции
Важно: Не забудьте привести энтропийный член к одинаковым единицам измерения! (ΔS° обычно в Дж/(моль·К), а ΔH° – в кДж/моль).
Учёт температурной зависимости
1. Температурная зависимость энтальпии (ΔH(T)):
2. Температурная зависимость энтропии (ΔS(T)):
3. Температурная зависимость энергии Гиббса (ΔG(T)) – ключевое уравнение:
Расчёт температуры начала реакции
Реакция становится термодинамически возможной при ΔG(T) ≤ 0. Температуру, при которой система находится в равновесии (ΔG = 0), находят из уравнения:
ΔH°р-ции-Tравн·ΔS°р-ции=0
Отсюда:
Tравн=ΔH°р-ции/ΔS°р-ции
Этот расчёт даёт ориентировочную температуру. Реакция будет самопроизвольно протекать при T > Tравн, если ΔS > 0, и при T < Tравн, если ΔS < 0.
Условия самопроизвольного протекания реакции в зависимости от знаков ΔH и ΔS:
Для эндотермических реакций (ΔH > 0):
Если энтропия возрастает (ΔS > 0), реакция становится возможной при T > Tравн. Преодоление энергетического барьера (ΔH > 0) происходит за счёт возрастания беспорядка в системе (ΔS > 0), что становится выгодно при высоких температурах.
Для экзотермических реакций (ΔH < 0):
Если энтропия убывает (ΔS < 0), реакция возможна при T < Tравн. Движущей силой реакции является выделение энергии (ΔH < 0), и этот эффект доминирует при низких температурах, пока не перевесит фактор уменьшения беспорядка.
Этот алгоритм позволяет на основе табличных данных сделать количественный прогноз о возможности и условиях протекания химической реакции. Помните, что отрицательное значение ΔG – это критерий принципиальной возможности, но не гарантия того, что реакция пойдёт с заметной скоростью.
Пример расчёта для реакции разложения карбоната кальция
Рассмотрим реакцию: CaCO₃(тв) → CaO(тв) + CO₂(г)
Цель: определить, при какой температуре начнется разложение CaCO₃ (т.е. когда ΔG(T) станет меньше нуля).
Шаг 1. Находим стандартные термодинамические данные в справочнике:
Кинетика ферментативных реакций: уравнение Михаэлиса–Ментена и линеаризация Лайнуивера–Берка
Краткое описание строения и свойств ферментов
Ферменты, или энзимы — это чаще всего молекулы белковой природы, катализирующие химические реакции в живых системах. Кроме белков, существует немногочисленная группа РНК ферментов.
Как и все белки, ферменты синтезируются изначально в виде полипептидной цепочки — первичная структура. Потом за счёт образования водородных связей и дисульфидных мостиков между между молекулами аминокислот в цепи образуется вторичная структура фермента. В дальнейшем под действием Ван-Вер-Вальсовых, электростатических и нековалентных сил образуется третичная и четвертичная структуры ферментов.
В каталитическом процессе участвует не весь фермент, а только его малая часть — активный центр.
Кроме этого стоит понимать, что любой фермент обладает высокой специфичностью и активностью. Для того чтобы катализировать реакцию, фермент связывается только с одним или несколькими определёнными субстратами. Большинство ферментативных процессов идёт со скоростями в 108-1020 раз большими, чем если бы они протекали сами. Говоря об активности ферментов, то их эффективность как катализаторов зачастую выше в 108-109 раз, чем их неорганические аналоги.
Кинетика ферментативных процессов
В основе кинетического анализа ферментативного процесса лежит предположение о том, что между ферментом (E) и субстратом (S) происходит обратимое образование фермент-субстратного комплекса. В дальнейшем в результате протекания реакции субстрат необратимо преобразуется в продукт (P) и комплекс распадается.
Используя допущение о стационарном протекании реакции, применим метод квазистационарных концентраций для фермент-субстратного комплекса.
Учтём тот факт, что фермент при реакции находится как в свободной форме, так и в форме фермент-субстратного комплекса. Выразим тогда его свободную форму и подставим в квазистационарное уравнение.
Выразим из этого уравнения [ES].
Скорость ферментативной реакции в основном определяется скоростью образования продукта реакции из субстрат ферментного комплекса. Тогда:
Разделим в получившемся уравнении и числитель, и знаменатель на K1 и получим:
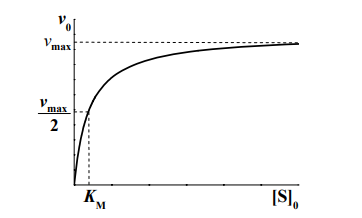
KM — это константа Михаэлиса, показывающая концентрацию субстрата, при которой скорость реакции равна половине максимальной. Численное значение KM зависит от многих параметров:
pH среды, где протекает реакции;
Температуры, при которой проводится реакция;
Концентрации реагента, который подвергается ферментативному преобразованию;
Концентрация продукта ферментирования;
Наличию или отсутствию активаторов/ингибиторов ферментов
В результате действиях всех этих факторов KM может меняться в приделах от 1 до 10-8 моль*л-1. Стоит также отметить, что чем меньше значение KM, тем больше сродство фермента к субстрату, и наоборот. Кроме этого, ферментативные реакции часто проводят в буферных растворах с контролируемой ионной силой, чтобы избежать изменения KM (это можно прочитать в конспекте Гусаровой А.Р. «Расчёт осмотического давления и других коллигативных свойств растворов»).
На начальных этапах, когда количество субстрата превышает количество фермента, а концентрация продукта стремиться к нулю, уменьшением концентрации субстрата можно пренебречь. Тогда:
Если же K2>>K3, то в начале реакции устанавливается квазистатическое равновесие и константа Михаэлиса заменяется на субстратную константу (KS).
Проанализируем уравнение для v0 с KM для двух предельных случаев:
[S]0 << KM, тогда реакция имеет первый порядок как по ферменту, так и по субстрату;
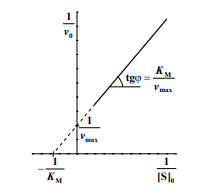
[S]0 >> KM, в этом случае начальная скорость реакции не зависит от концентрации субстрата и называется максимальной скоростью ферментативной реакции vmax .Этот эффект так называемого субстратного насыщения обусловлен практически полным связыванием всего имеющегося в системе фермента в фермент-субстратный комплекс, поэтому его концентрация, а, следовательно, и наблюдаемая скорость реакции перестают зависеть от концентрации субстрата. Тогда
Это уравнение было получено Бриггсом и Холдейном в 1925 г., но названо ими уравнением Михаэлиса−Ментена. На практике же, невозможно никогда достичь vmax, так как работа фермента очень сильно зависит и от концентрации продукта реакции. Чем больше концентрация продукта, тем меньше сродство фермента к субстрату. Следует также помнить, что начальная скорость ферментативной реакции часто определяется по оптической плотности (это можно прочитать в конспекте Борисова В.В. «Построение градуировочного графика в спектрофотометрии и его статистическая обработка»).
На рисунке представлена зависимость начальной скорости ферментативной реакции от начальной концентрации субстрата.
Стоит обратить внимание, что построение такого графика для определения vmax и KM на практике очень сложно. Поэтому для определения KM и vmax графически используют преобразованное уравнение Михаэлиса-Ментена, предложенное Г.Лайнуивером и Д.Бэрком для линеаризации графика.
Графическое определение KM и vmax в координатах Лайнуивера-Берка
Максимальную скорость можно определить по отрезку, отсекаемому на оси ординат, 1/vmax , а константу Михаэлиса можно вычислить либо по тангенсу угла наклона прямой, равному KM/vmax , либо по отрицательному отрезку, отсекаемому на оси абсцисс.
Недостатки модели Михаэлиса
Несмотря на способность модели Михаэлиса-Ментена к упрощению анализа ферментативных процессов, её применение сопряжено с рядом методологических и практических ограничений.
Уравнение можно применять только при стационарных условиях;
Недопущение наличия продуктов ферментативной реакции в начальной системе;
Не подходит для определения скорости реакции в реакциях с аллостерическими ферментами.
Заключение и выводы
Ферменты — это сложные белковые молекулы, представляющие из себя биокатализаторы;
Константа Михаэлиса — это константа, показывающая концентрацию субстрата, при которой скорость реакции равна половине максимальной;
Уравнением Михаэлиса−Ментена имеет очень много допущений, вследствие чего теоретические значения, полученные при расчёте, могут сильно отличаться от полученных экспериментально;
График получаемый при решении уравнения Михаэлиса−Ментена очень сложен для определения KM и vmax на практике, поэтому используют линеаризированное уравнение, предложенное Г.Лайнуивером и Д.Бэрком.
Пример решения задачина уравнение Михаэлиса-Ментена
Фермент катализирует превращение субстрата S в продукт P по кинетике Михаэлиса–Ментена. При Vmax = 120 нмоль/мин и KM = 30 мкМ найдите начальную скорость реакции при концентрациях субстрата:
[S]0 = 3 мкМ;
[S]0 = 30 мкМ;
[S]0 = 300 мкМ;
Решение задачи
Воспользуемся уравнением Михаэлиса-Ментена и подставим известные нам величины:
Следует помнить, что при [S]0 ≪ Km начальная скорость пропорциональна [S]; при [S]0 = Km начальная скорость равна половине Vmax; при [S]0 ≫ Km начальная скорость стремится к Vmax.
Коллигативные свойства растворов и осмотическое давление
Природа коллигативных свойств
Коллигативные свойства — это совокупность физико-химических свойств разбавленных растворов, которые зависят только от молярной доли (или концентрации) растворенного вещества и не зависят от его химической природы.
Эти свойства являются следствием понижения химического потенциала растворителя при введении в него нелетучего растворенного вещества. Раствор стремится к состоянию с более высоким химическим потенциалом, что и проявляется в наблюдаемых эффектах.
Условия применимости:
Раствор должен быть идеальным или достаточно разбавленным.
Растворенное вещество — нелетучее.
Растворенное вещество не диссоциирует и не ассоциирует (для базовых формул).
Основные коллигативные свойства и их закономерности
Относительное понижение давления пара растворителя над раствором равно молярной доле растворенного вещества.
(P₀ — P) / P₀ = χ₂ = n₂ / (n₁ + n₂)
P₀ — давление пара чистого растворителя [Па].
P — давление пара над раствором [Па].
χ₂ — молярная доля растворенного вещества.
n₁, n₂ — количество моль растворителя и растворенного вещества соответственно [моль].
Выведем формулу, используемую в сильно разбавленных растворах.Учтем,что в сильно-разбавленных растворах количество растворителя превышает количество растворенного вещества, значит n1=0, тогда
P₀ ≈ n₂ / n₁ = M₁ * m / 1000
m — моляльность [моль/кг],
M₁ — молярная масса растворителя [г/моль]
Кроме того, следует помнить, что существует азеотропные растворы, отклоняющиеся от закона Рауля по причинам, которые можно узнать в следующем конспекте
Понижение давления пара приводит к необходимости нагревания раствора до более высокой температуры для достижения давления, равного атмосферному.
Kэ — эбуллиоскопическая константа. Для воды Kэ = 0,516 [К·кг/моль].
m — моляльная концентрация [моль/кг].Использование моляльной концентрации в этом уравнении связано с тем, что в других способах выражения концентраций количество растворителя зависит от количества растворённого вещества, а в моляльной концентрации количество растворителя фиксировано (1000 граммов).
i — изотонический коэффициент Вант-Гоффа.
Понижение температуры замерзания (Криоскопия)
Наличие примеси нарушает кристаллическую решетку растворителя, требуя большего переохлаждения для начала кристаллизации.
ΔTзам = Tзам(р-ля) — Tзам(р-ра) = Kк · m · i
Kк — криоскопическая константа. Для воды Kк = 1,86 [К·кг/моль].
m — моляльная концентрация [кг/моль] .
Оба явления прямо пропорциональны моляльной концентрации и описываются схожими уравнениями, но с разными физическими константами растворителя.
Осмотическое давление:
Осмос — направленная диффузия молекул растворителя через полупроницаемую мембрану из области с меньшей концентрацией растворенного вещества в область с большей его концентрацией.
Осмотическое давление (π) — это избыточное гидростатическое давление, которое необходимо приложить к раствору, чтобы предотвратить его разбавление чистым растворителем через полупроницаемую мембрану. Это коллигативное свойство, наиболее чувствительное к концентрации и потому особенно важное для изучения высокомолекулярных соединений.
Уравнение Вант-Гоффа и его вывод
Уравнение выводится из равенства химических потенциалов растворителя по обе стороны мембраны в условиях равновесия.
π = i · C · R · T
π — осмотическое давление [кПа, атм].
i — изотонический коэффициент. Показывает, во сколько раз общее число частиц в растворе больше числа исходных молекул из-за диссоциации (i = 1 + α(k-1), где α — степень диссоциации, k — число ионов из одной молекулы).
C — молярная концентрация растворенного вещества [моль/м³ или моль/л]. Важно: для точных расчетов использовать СИ (моль/м³).
R — универсальная газовая постоянная (8,314 Дж/(моль·К)).
T — абсолютная температура [K].
Коэффициент i связывает теорию идеальных растворов с поведением реальных растворов, особенно электролитов.
В реальных концентрированных растворах i может отличаться от теоретического из-за межионных взаимодействий.
Для неэлектролитов (глюкоза, сахароза)
i = 1
Для сильных электролитов (NaCl, KCl)
NaCl → Na⁺ + Cl⁻ (k=2), теоретически i ≈ 2 CaCl₂ → Ca²⁺ + 2Cl⁻ (k=3), теоретически i ≈ 3
Очевидно, что взаимодействие ионов уменьшается с повышением температуры, а также с уменьшением их концентрации, то есть, разбавлением раствора, ведь тогда уменьшается вероятность встречи двух частичек.Об этом подробнее можно узнать в
Рассчитайте осмотическое давление при 37°C раствора, содержащего 4,0 г гидроксида натрия (NaOH) и 18,0 г глюкозы (C₆H₁₂O₆) в 500 мл воды. Плотность раствора принять равной 1 г/мл. Считать NaOH сильным электролитом.
Дано:
m(NaOH) = 4,0 г
m(глюкозы) = 18,0 г
V(р-ра) ≈ 0,5 л (по условию, т.к. плотность ~1 г/мл)
T = 37°C = 310 K
R = 8,31 Дж/(моль·К) = 8,31 кПа·л/(моль·К)
M(NaOH) = 40 г/моль
M(глюкозы) = 180 г/моль
i(NaOH) ≈ 2 (NaOH → Na⁺ + OH⁻)
i(глюкозы) = 1
Решение:
1. Рассчитаем количество вещества и концентрацию для каждого компонента.
Для NaOH:
n(NaOH) = m / M = 4,0 г / 40 г/моль = 0,1 моль
C(NaOH) = n / V = 0,1 моль / 0,5 л = 0,2 М
С учетом диссоциации: Cэфф(NaOH) = i · C = 2 · 0,2 М = 0,4 М
Для глюкозы:
n(глюкозы) = 18,0 г / 180 г/моль = 0,1 моль
C(глюкозы) = 0,1 моль / 0,5 л = 0,2 М
Cэфф(глюкозы) = i · C = 1 · 0,2 М = 0,2 М
2. Найдем общую эффективную молярную концентрацию частиц в растворе.
Cобщ = Cэфф(NaOH) + Cэфф(глюкозы) = 0,4 М + 0,2 М = 0,6 моль/л
3. Рассчитаем осмотическое давление по уравнению Вант-Гоффа.
π = Cобщ · R · T
π = 0,6 моль/л · 8,31 кПа·л/(моль·К) · 310 K
π ≈ 1546 кПа
Ответ: Осмотическое давление раствора составляет 1546 кПа или около 15,3 атм.
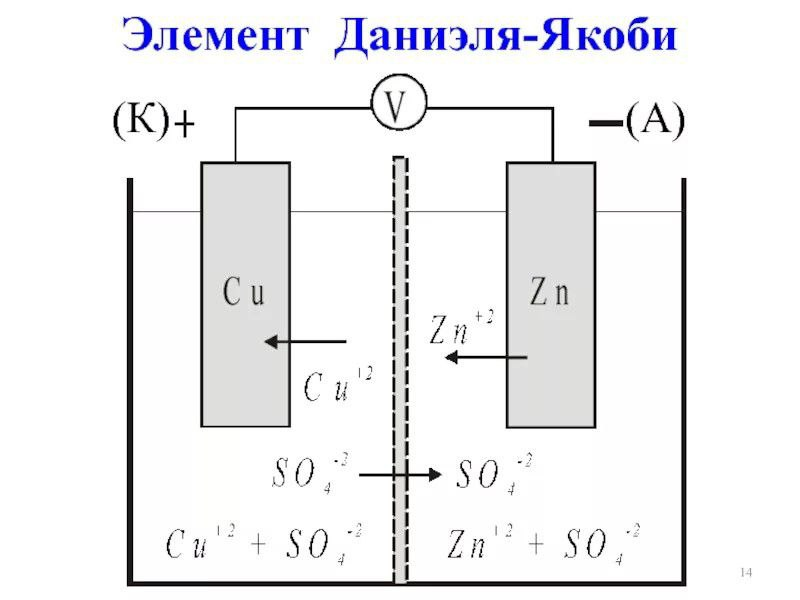
Расчет ЭДС гальванического элемента и определение направления реакции
1. Введение: Что такое гальванический элемент?
Гальванический элемент — это устройство, которое генерирует электричество за счёт химической реакции. Представь батарейку: внутри неё металл окисляется, а другой металл восстанавливается.
Пример: Даниеллев элемент (медь и цинк). Два электрода (анод и катод) и электролит. Анод — где происходит окисление (потеря электронов), катод — где восстановление (приобретение электронов).
2. Теория: Расчёт ЭДС
ЭДС (Е°) — это разность потенциалов между анодом и катодом. Она показывает, насколько «хочет» идти реакция или идёт ли она в прямом направлении или в обратном.
2.1. Стандартная ЭДС
Формула: Е° клетки =Е°_ восст(катода) — Е°_восст( анода)
Значения берутся из таблицы стандартных потенциалов (например, Zn²⁺/Zn = -0.76 B, Cu²⁺/Cu = +0.34 B.)
Все потенциалы, приведенные в таблице, являются потенциалами восстановления. Табличные E° — для водных растворов, 25 °C, 1 М (или 1 бар для газов). Реальные элементы могут иметь поляризацию, перенапряжение — но это выходит за рамки базового курса.
Пример: Даниеллев элемент:
Е° катода (Cu²⁺/Cu) = +0.34 В.
Е°анода (Zn²⁺/Zn) = -0.76 В.
Е° клетки = 0.34 — (-0.76) = 1.10 В.
ЭДС = 1.10 В
2.2. Уравнение Нернста (для нестандартных условий)
Формула: Е = Е° — (RT / nF) * In(Q)
Е — ЭДС при данных условиях.
Е° — стандартная ЭДС.
R = 8.31 Дж/(моль-К) (газовая постоянная).
Т — температура (в К, например, 298 К для 25 °C).
n — число передаваемых электронов.
F = 96485 Кл/моль (Фарадей).
Q — реакционная квота (соотношение концентраций продуктов к реагентам)
Построение градуировочного графика в спектрофотометрии и его статистическая обработка
1. Теоретические основы
1.1. Теория Метода градуировочного графика
В аналитической химии для определения концентрации данного вещества в растворе помимо титрометрии, гравиметрии пользуются также спектрофотометрией. Одним из методов количественной оценки анализируемого вещества является Метод Градуировочного Графика.
В этом методе готовят серию стандартных окрашенных растворов концентрации которых охватывают область возможных изменений концентрации анализируемого раствора и строят график зависимости измеряемой оптической плотности (A) от концентрации вещества (C) в серии стандартных растворов.
Наиболее эффективно этот метод показывает себя при многократном фотометрировании однотипных по химическому составу растворов.
Основополагающий закон, согласно которому оптическая плотность прямо пропорциональна концентрации – закон Бугера-Ламберта-Бера (БЛБ):
A =E × l × C
A – оптическая плотность раствора (изменение интенсивности света на выходе из анализируемого раствора)
Е – молярный коэффициент поглощения (показывает способность данного вещества поглощать свет определённой длины)
l – путь, преодолеваемый лучом света через анализируемый раствор
С – концетрация данного вещества в анализируемом растворе
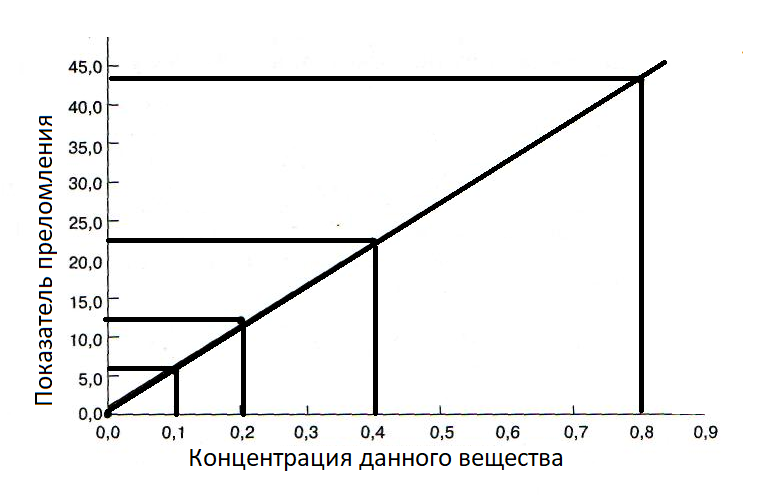
Пример градуировочного графика:
В спектрометрах путь постоянен и равен длине кюветы. Молярный коэфициент поглощения зависит от длины волны и постоянен при монохроматическом излучении. Поэтому зависимость A от C в идеальной модели – линейная, но из-за примесей в растворе, сложности выполнения монохроматичности волны, приборной ошибки приемника и т.д. могут наблюдаться отклонения.
1.2. Теория Метода Наименьших квадратов.
Для учёта отклонений от линейной зависимости пользуются различными математическими методами, в данном конспекте описывается Метод Наименьших Квадратов.
Метод наименьших квадратов (МНК) используется для нахождения уравнения линейной зависимости (y = ax + b), которая наилучшим образом отражает экспериментальные данные. Задача МНК сводится к тому, чтобы найти такие значения коэффициентов «a» и «b», при которых сумма квадратов расстояний точек снизу от прямой, равна сумме квадратов расстояний точек.
Эти коэффициенты легче всего найти, пользуясь специализируемыми программами обработки данных, например, Excel.
Иначе, пользуются формулами:
а – тангенс угла наклона графика относительно оси Х. b — отрезок, отсекаемый графиком на оси Y. Xi — концентрация анализируемого вещества в образце. Y – Оптическая плотность. n — число точек
2. Пошаговый алгоритм
Приготовление стандартных растворов: Готовится серия растворов с известной, возрастающей концентрацией определяемого вещества. Каждой концентрации по несколько образцов. Это нужно для оценки воспроизводимости — т.е. насколько значения оптической плотности отличаются в разных растворах при одинаковой концентрации определяемого вещества в растворах.
Измерение сигнала: Сначала измеряют показатель оптической плотности для раствора, который содержит все компоненты анализируемого образца, кроме самого определяемого вещества. Такой опыт называется “Холостым” (blanc), полученное значение принимают за ноль и дальнейшие замеры будут отражать повышение оптической плотности только из-за наличия в растворе определяемого вещества. Далее на спектрофотометре измеряется оптическая плотность (A) каждого стандартного раствора при выбранной длине волны.
Построение графика: На график наносятся точки: по оси X – концентрация (C, моль/л), по оси Y – оптическая плотность (A).
Статистическая обработка (МНК): Рассчитывается показатели постоянных «а» и «b» для линейной зависимости.
Расчет концентрации неизвестного образца: Измеряется оптическая плотность образца (Ax). Его концентрация (Cx) рассчитывается по уравнению калибровочного графика:
3. Разбор примера
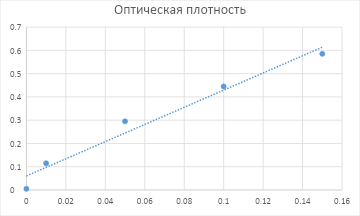
1-2) Приготовили ряд стандартных растворов для соединения K2CrO4 и измерили оптическую плотность для каждого раствора. Получили следующие данные:
Концентрация определяемого вещества в растворе, моль/л
Оптическая плотность
0,0
0,000
0,01
0,115
0,05
0,295
0,1
0,445
0,15
0,585
3-4) Построим график из полученных данных:
Далее, с помощью МНК (вручную или в программе, напр., Excel) получим уравнение:
A = 3,6877 * C + 0.0604
5) Допустим анализируемый раствор дал значение A = 0.350.


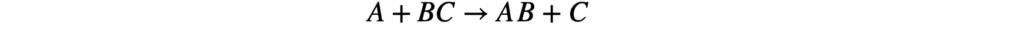



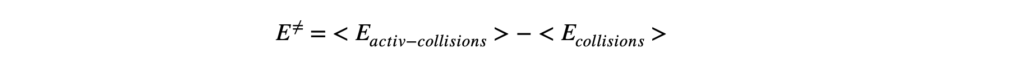
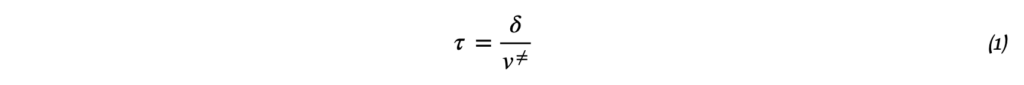
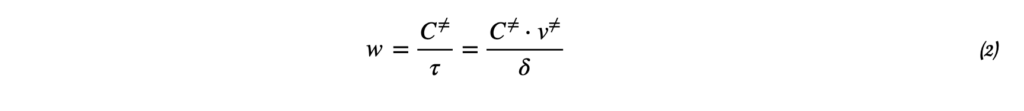
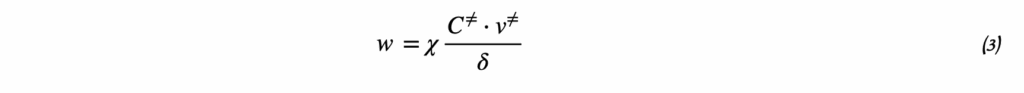
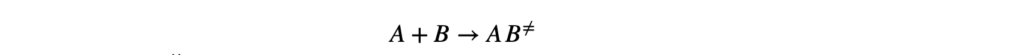
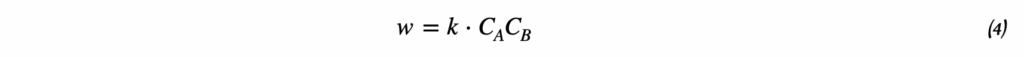
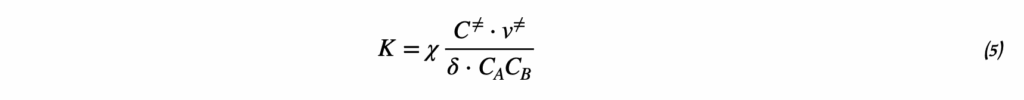

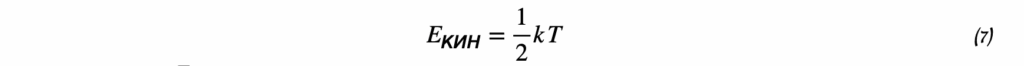
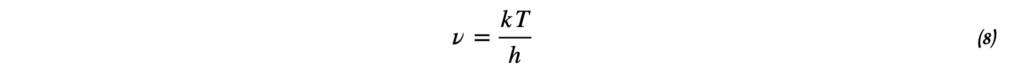
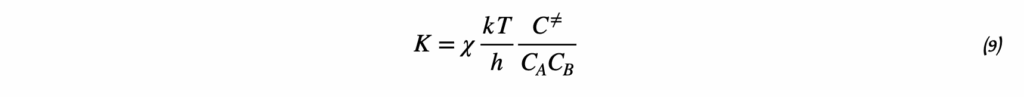
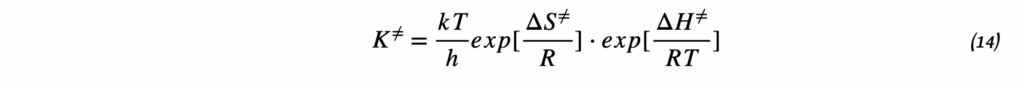
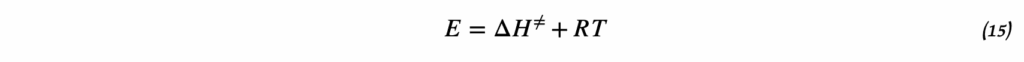

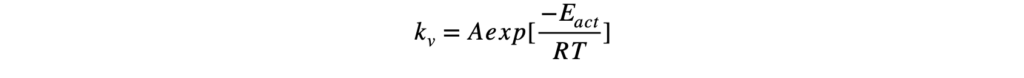
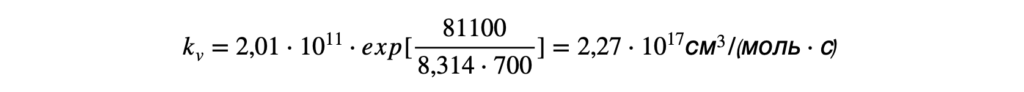
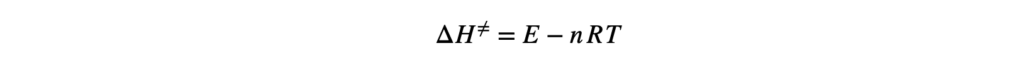
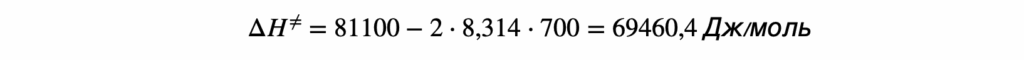
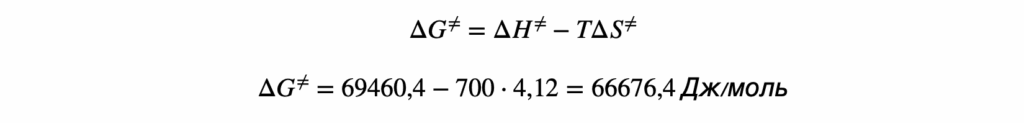
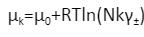
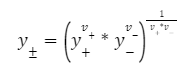
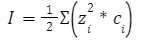
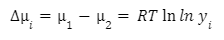
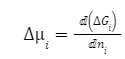
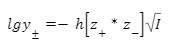
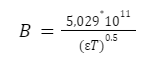
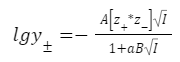
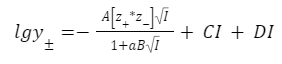

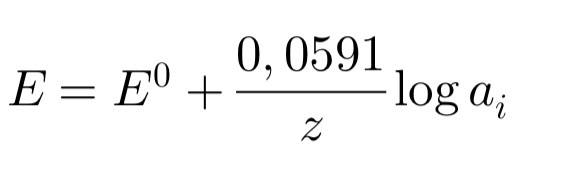


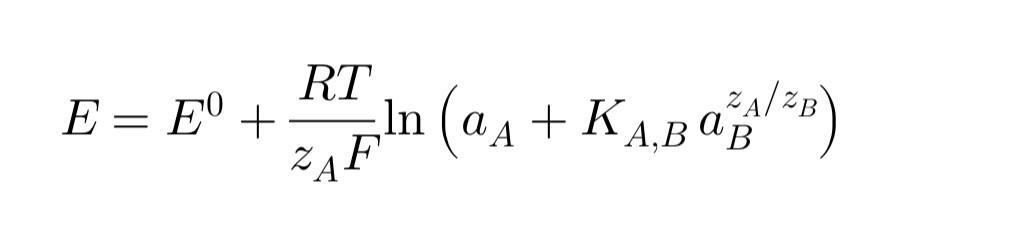
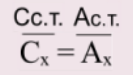
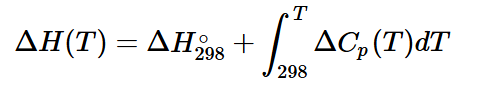
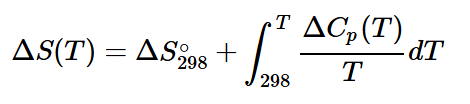
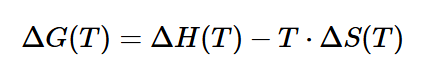
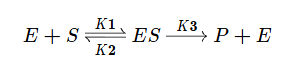
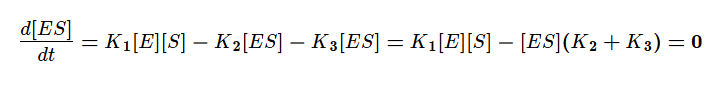
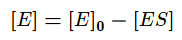
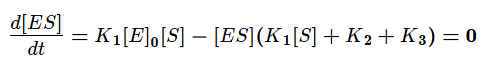
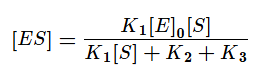
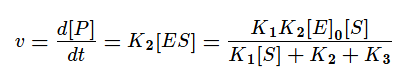
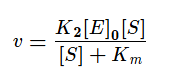
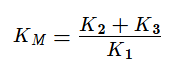
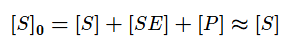
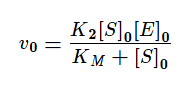
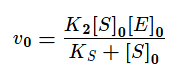
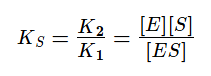


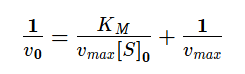


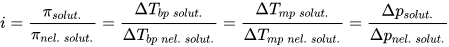
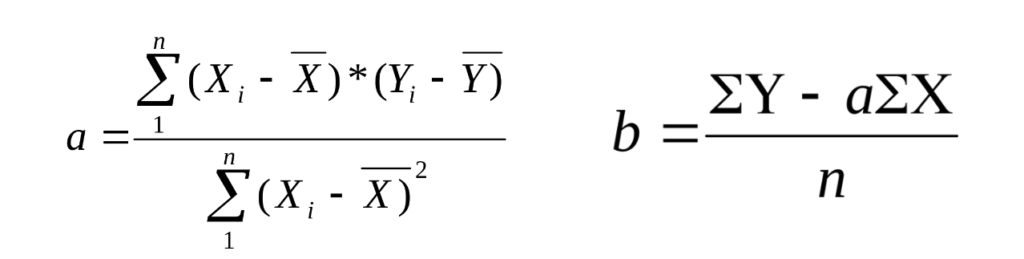
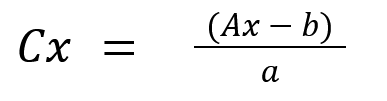
Новости
22.11.2024
22.11.2024
24.10.2024
14.10.2024
09.10.2024
01.10.2024