Построение градуировочного графика в спектрофотометрии и его статистическая обработка
1. Теоретические основы
1.1. Теория Метода градуировочного графика
В аналитической химии для определения концентрации данного вещества в растворе помимо титрометрии, гравиметрии пользуются также спектрофотометрией. Одним из методов количественной оценки анализируемого вещества является Метод Градуировочного Графика.
В этом методе готовят серию стандартных окрашенных растворов концентрации которых охватывают область возможных изменений концентрации анализируемого раствора и строят график зависимости измеряемой оптической плотности (A) от концентрации вещества (C) в серии стандартных растворов.
Наиболее эффективно этот метод показывает себя при многократном фотометрировании однотипных по химическому составу растворов.
Основополагающий закон, согласно которому оптическая плотность прямо пропорциональна концентрации – закон Бугера-Ламберта-Бера (БЛБ):
A =E × l × C
A – оптическая плотность раствора (изменение интенсивности света на выходе из анализируемого раствора)
Е – молярный коэффициент поглощения (показывает способность данного вещества поглощать свет определённой длины)
l – путь, преодолеваемый лучом света через анализируемый раствор
С – концетрация данного вещества в анализируемом растворе
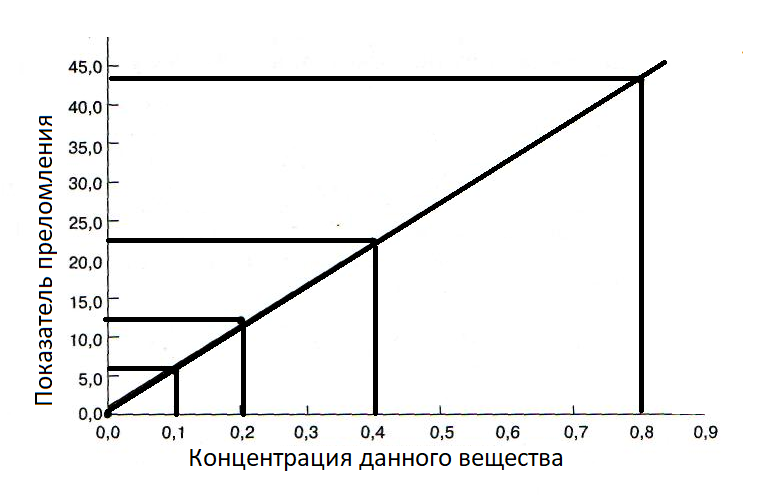
Пример градуировочного графика:
В спектрометрах путь постоянен и равен длине кюветы. Молярный коэфициент поглощения зависит от длины волны и постоянен при монохроматическом излучении. Поэтому зависимость A от C в идеальной модели – линейная, но из-за примесей в растворе, сложности выполнения монохроматичности волны, приборной ошибки приемника и т.д. могут наблюдаться отклонения.
1.2. Теория Метода Наименьших квадратов.
Для учёта отклонений от линейной зависимости пользуются различными математическими методами, в данном конспекте описывается Метод Наименьших Квадратов.
Метод наименьших квадратов (МНК) используется для нахождения уравнения линейной зависимости (y = ax + b), которая наилучшим образом отражает экспериментальные данные. Задача МНК сводится к тому, чтобы найти такие значения коэффициентов «a» и «b», при которых сумма квадратов расстояний точек снизу от прямой, равна сумме квадратов расстояний точек.
Эти коэффициенты легче всего найти, пользуясь специализируемыми программами обработки данных, например, Excel.
Иначе, пользуются формулами:
а – тангенс угла наклона графика относительно оси Х. b — отрезок, отсекаемый графиком на оси Y. Xi — концентрация анализируемого вещества в образце. Y – Оптическая плотность. n — число точек
2. Пошаговый алгоритм
Приготовление стандартных растворов: Готовится серия растворов с известной, возрастающей концентрацией определяемого вещества. Каждой концентрации по несколько образцов. Это нужно для оценки воспроизводимости — т.е. насколько значения оптической плотности отличаются в разных растворах при одинаковой концентрации определяемого вещества в растворах.
Измерение сигнала: Сначала измеряют показатель оптической плотности для раствора, который содержит все компоненты анализируемого образца, кроме самого определяемого вещества. Такой опыт называется “Холостым” (blanc), полученное значение принимают за ноль и дальнейшие замеры будут отражать повышение оптической плотности только из-за наличия в растворе определяемого вещества. Далее на спектрофотометре измеряется оптическая плотность (A) каждого стандартного раствора при выбранной длине волны.
Построение графика: На график наносятся точки: по оси X – концентрация (C, моль/л), по оси Y – оптическая плотность (A).
Статистическая обработка (МНК): Рассчитывается показатели постоянных «а» и «b» для линейной зависимости.
Расчет концентрации неизвестного образца: Измеряется оптическая плотность образца (Ax). Его концентрация (Cx) рассчитывается по уравнению калибровочного графика:
3. Разбор примера
1-2) Приготовили ряд стандартных растворов для соединения K2CrO4 и измерили оптическую плотность для каждого раствора. Получили следующие данные:
Концентрация определяемого вещества в растворе, моль/л
Оптическая плотность
0,0
0,000
0,01
0,115
0,05
0,295
0,1
0,445
0,15
0,585
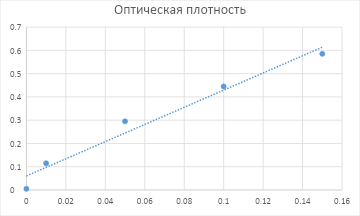
3-4) Построим график из полученных данных:
Далее, с помощью МНК (вручную или в программе, напр., Excel) получим уравнение:
A = 3,6877 * C + 0.0604
5) Допустим анализируемый раствор дал значение A = 0.350.
Гетерогенная система – система, в составе которой присутствуют две или более однородных части (фазы), имеющие четкую поверхность раздела. Эти фазы могут различаться между собой по своим физическим свойствам и химическому составу.
Фаза – это однородная составляющая гетерогенной системы, которая характеризуется постоянством химического состава и физических свойств в любой своей точке. Она отделена от других фаз видимой границей раздела и рассматривается применительно к макроскопическим, а не к молекулярным объемам.
Составляющее вещество системы – каждое химическое вещество, которое может быть выделено из системы и существовать в изолированном виде.
Компонент (к) – независимая составляющая системы, минимальное число химических индивидуальных веществ, необходимых для выражения состава всех существующих в системе фаз. Для систем, где не идут химические реакции, число компонентов обычно равно числу составляющих веществ.
Число степеней свободы (f) – количество независимых параметров состояния (таких как температура, давление, концентрация компонентов), которые можно произвольно изменять в некоторых пределах, не нарушая равновесия и не изменяя числа фаз в системе.
2. Вывод правила фаз Гиббса
Рассмотрим равновесную гетерогенную систему, состоящую из Ф фаз, в каждую из которых входят все k компонентов. В условиях равновесия температура (T) и давление (P) одинаковы во всех фазах, а химический потенциал каждого компонента также имеет одинаковое значение во всех фазах.
Чтобы описать состав одной фазы, содержащей k компонентов, достаточно задать концентрации (k-1) компонента. Следовательно, для определения состава всех Ф фаз потребуется Ф(k-1) переменных концентраций. Добавляя к ним два внешних параметра (температуру и давление), получаем общее число переменных: Ф(k-1) + 2.
Однако эти переменные не являются независимыми. На них накладываются условия равновесия, которые выражаются через равенство химических потенциалов для каждого компонента по всем фазам.
Для одного компонента это дает (Ф — 1) независимое уравнение. Для k компонентов общее число таких уравнений связи составит k(Ф — 1).
Таким образом, число степеней свободы (f), то есть число действительно независимых переменных, равно общей численности переменных за вычетом числа уравнений связи между ними:
f = [Ф(k-1) + 2] — k(Ф — 1)
Раскрыв скобки и упростив выражение, получаем:
f = k — Ф + 2
3. Формулировка правила фаз и его модификации
f = k — Ф + 2
Число степеней свободы равновесной гетерогенной термодинамической системы, на которую из внешних факторов влияют только температура и давление, равно числу компонентов системы минус число фаз плюс два.
ПРАВИЛО ФАЗ ГИББСА
Правило фаз может быть модифицировано, если на систему действуют иные факторы:
● Если добавляется еще один внешний параметр, одинаковый для всех фаз (например, электрический потенциал), то правило принимает вид:
f = k — Ф + 3.
● Если же один из параметров фиксирован (например, давление постоянно), то формула упрощается:
f = k — Ф + 1.
4. Ограничения правила фаз
Важно помнить, что правило фаз Гиббса применимо с рядом ограничений:
● Оно справедливо только для систем, находящихся в состоянии истинного термодинамического равновесия.
● Оно не учитывает влияние таких факторов, как гравитационные, магнитные или электрические поля (если только они не добавлены в формулу явно), а также поверхностные натяжения на границах раздела фаз.
● Предполагается, что фазы являются изотропными и их свойства не зависят от направления.
5. Пример применения правила фаз к воде
Вода в однокомпонентной системе (k=1) может существовать в виде нескольких фаз: различных кристаллических модификаций льда, жидкой воды и пара. Правило фаз для такой системы:
f = 3 — Ф.
● Внутри одной фазы (например, область жидкости на диаграмме состояния, точка a): Ф=1, следовательно, f=2. Система бивариантна – можно одновременно и независимо изменять и температуру, и давление, не изменяя числа фаз.
𝑓 = 𝑛 + 2 − Ф = 1 + 2 − 1 = 2
● На линиях равновесия между двумя фазами (например, линия «жидкость-пар», точка b): Ф=2, f=1. Система моновариантна – можно изменять только один параметр (либо температуру, либо давление), в то время как второй будет определяться однозначно по кривой равновесия.
𝑓 = 1 + 2 − 2 = 1
● В тройной точке O (лед-вода-пар): Ф=3, f=0. Система инвариантна – ни температуру, ни давление нельзя изменить, не вызвая исчезновения одной из фаз. Это состояние фазового равновесия строго определено.
f=1+2-3=0
Диаграмма состояния воды наглядно отображает эти области:
● OK – кривая давления насыщенного пара над жидкостью.
● OA – кривая зависимости температуры плавления льда от давления.
● OB – кривая давления пара над льдом.
● O – тройная точка.
● OC – кривая метастабильного равновесия пара и переохлажденной воды.
Правило фаз подтверждает, что максимальное число фаз, которые могут одновременно сосуществовать в равновесии в однокомпонентной системе, равно трем (f=0).
«Правило фаз» — при анализе электрохимических систем с осадками (например, AgCl) число фаз влияет на равновесие.
Температурная зависимость константы скорости. Уравнение Аррениуса и определение энергии активации.
1. Введение: Почему температура так важна?
Из повседневного опыта мы знаем, что скорость большинства химических реакций увеличивается с ростом температуры. Пища быстрее готовится, металлы быстрее ржавеют, а биологические процессы ускоряются в тепле. Количественно эта зависимость описывается константой скорости (k), которая входит в основное кинетическое уравнение.Задача химической кинетики — найти математическое выражение, которое точно описывало бы зависимость k = f(T). Шведский ученый Сванте Аррениус в 1889 году предложил наиболее успешную теорию.
2. Эмпирическая форма уравнения Аррениуса.
На основе критического анализа и переосмысления уже существовавших экспериментальных данных (гидролиз сахарозы, разложение N₂O₅), а также под влиянием его собственных работ по электролитической диссоциации. Аррениус перевел эмпирическое наблюдение в математический закон:
k=A*e^-(Ea/RT)
где:
Переменная
Определение
В чём выражается
k
Константа скорости реакции
Для реакции нулевого порядка: k = моль/(литр · секунда) или М/с (моль/л · с)| Для реакции первого порядка: k = 1/секунда или с⁻¹ Для реакции второго порядка: k = литр/(моль · секунда) или М⁻¹·с⁻¹ (л/моль · с) Для реакции n-го порядка: Общая формула: k = (концентрация)¹⁻ⁿ / время
A
Предэкспоненциальный множитель, отражает общее число столкновений частиц в единицу времени
Совпадает с константой K
e
Основание натурального логарифма
~2.718
Ea
Энергия активации
кДж/моль
R
Универсальная газовая постоянная
8.314 Дж/(моль·К
T
Абсолютная температура
Кельвин
Физический смысл уравнения: Константа скорости k определяется произведением общего числа столкновений (A) на долю эффективных столкновений
3. Физическая интерпретация: Энергия активации и теория активных соударений.
Для объяснения экспоненциального характера зависимости, Аррениус ввел понятие энергии активации (Eₐ). Энергия активации — это избыточная энергия, которой должны обладать частицы реагентов по сравнению со средней энергией, чтобы столкновение между ними привело к химической реакции.
Реагенты находятся в «долине» до горки.
Продукты — в «долине» после горки.
Энергия активации Eₐ — это высота горки, которую нужно преодолеть.
Чем выше горка (Eₐ), тем меньше частиц сможет ее преодолеть при данной температуре, и тем медленнее идет реакция.
3.1. Распределение Максвелла-Больцмана
Молекулы в системе движутся с разными скоростями и, следовательно, обладают разной кинетической энергией. Распределение Максвелла-Больцмана описывает долю молекул, обладающих энергией, равной или превышающей некоторое значение.
e^-(Ea/RT)
Доля частиц с энергией ≥ Eₐ пропорциональна множителю.
Как температура влияет на долю активных частиц:
При T₁ (низкая температура) доля частиц с E ≥ Eₐ невелика (площадь под кривой справа от Eₐ). Реакция идет медленно.
При T₂ (высокая температура) эта доля значительно возрастает. Именно поэтому небольшое увеличение температуры приводит к резкому росту скорости реакции — зависимость экспоненциальная.
Стерический фактор (p) — вероятность того, что столкновение произойдет с правильной пространственной ориентацией молекул. Даже если энергия достаточна, молекулы могут не прореагировать, если столкнутся «не теми» частями.
Таким образом, A = p · Z, где Z — общая частота столкновений.
4. Аналитическая форма уравнения Аррениуса и определение Eₐ
Экспоненциальная форма не всегда удобна для расчетов. Прологарифмируем уравнение Аррениуса:
Шаг 1: Логарифмирование
Шаг 2: Получение линейного уравнения Это уравнение имеет вид линейной функции y = b + a · x:
y = ln k
x = 1/T
a = -Ea / R (тангенс угла наклона прямой)
b = ln A (отрезок, отсекаемый на оси y)
Эта форма чрезвычайно полезна, так как позволяет определить Eₐ и A из экспериментальных данных.
Шаг 3: Экспериментальное определение Eₐ
Проводят серию экспериментов по измерению константы скорости k при разных температурах T.
Строят график в координатах ln k от 1/T (так называемый график Аррениуса).
Если экспериментальные точки накладываются на прямую линию (что выполняется для большинства реакций), это подтверждает справедливость уравнения Аррениуса для данной реакции.
Реакция разложения: 2H₂O₂ → 2H₂O + O₂ Без катализатора эта реакция идет очень медленно, так как имеет высокий энергетический барьер. В присутствии иодид-иона (I⁻) скорость реакции резко возрастает. Механизм этого катализа выглядит следующим образом:
Медленная стадия: H₂O₂ + I⁻ → H₂O + IO⁻ (константа скорости k₁, энергия активации Eₐ₁)
Быстрая стадия: H₂O₂ + IO⁻ → H₂O + O₂ + I⁻ (константа скорости k₂, энергия активации Eₐ₂)
Иодид-ион не расходуется, он является катализатором. Лимитирующей (определяющей общую скорость) является первая стадия.Экспериментально установлено, что кинетическое уравнение для этой реакции имеет вид:
v = k_набл [H₂O₂] [I⁻]
Как связана наблюдаемая константа скорости с константами отдельных стадий? В данном случае, для предлагаемого механизма, скорость образования продуктов определяется скоростью первой стадии:
v = k₁ [H₂O₂] [I⁻]
Следовательно, наблюдаемая константа скорости равна константе скорости первой стадии:
k_набл = k₁
Вывод для этого случая: Кажущаяся энергия активации Eₐ каж будет равна истинной энергии активации лимитирующей стадии: Eₐ каж = Eₐ₁. Катализатор ( I⁻ ) предоставляет альтернативный путь реакции с более низкой Eₐ₁ по сравнению с некатализируемой реакцией.
Реакции с нулевой энергией активации: Некоторые реакции (например, ионные в растворе) идут очень быстро и слабо зависят от температуры. Для них Eₐ ≈ 0.
Многостадийные сложные реакции: Для них график Аррениуса может не быть строго линейным, так как наблюдаемая константа скорости является комбинацией констант отдельных стадий, каждая со своей Eₐ.
Туннельные эффекты: Для очень легких частиц (электроны, протоны) при низких температурах становится существенным квантовомеханическое туннелирование через энергетический барьер, что не описывается классическим уравнением Аррениуса
7. Заключение и выводы:
Уравнение Аррениуса — фундаментальный закон химической кинетики, устанавливающий экспоненциальную зависимость константы скорости от температуры.
Энергия активация (Eₐ) — это минимальная избыточная энергия, необходимая для протекания реакции. Она является мерой «высоты» энергетического барьера реакции.
Предэкспоненциальный множитель (A) характеризует частоту и правильность ориентации столкновений частиц.
Линейная форма уравнения ln k = ln A — Eₐ/(RT) позволяет экспериментально определять Eₐ и A по данным о скорости реакции при разных температурах.
Знание Eₐ позволяет не только предсказывать скорость реакции при различных температурах, но и делать выводы о механизме реакции, что является одной из центральных задач физической химии.
Построение диаграммы распределения для полипротонных кислот
1. Основные понятия
Полипротонная кислота – это кислота, способная отдавать более одного протона (иона H⁺). Примеры: H₂SO₄, H₃PO₄, H₂CO₃, H₂C₂O₄.
Диаграмма распределения – это график, который показывает, как доля (α) каждой частицы (кислоты и продуктов её диссоциации) в растворе зависит от pH.
Доля (α) – безразмерная величина, показывающая мольную долю данной формы в общем аналитическом количестве кислоты. Сумма всех долей в любой точке равна 1 (или 100%).
2. Процесс диссоциации полипротонной кислоты
Рассмотрим трёхосновную кислоту H₃PO4. Её ступенчатая диссоциация описывается следующими уравнениями и константами:
H₃PO4 ⇄ H⁺ + H₂PO4⁻ Константа диссоциации: Ka₁
H₂PO4⁻ ⇄ H⁺ + HPO4²⁻ Константа диссоциации: Ka₂
HPO4²⁻ ⇄ H⁺ + PO4³⁻ Константа диссоциации: Ka₃
Для кислоты справедливо: Ka₁ > Ka₂ > Ka₃
Далее для удобства, в качестве общего виды кислоты будем иcпользовать H3A.
3. Формулы для расчёта долей форм
Общая аналитическая концентрация кислоты: C = [H₃A] + [H₂A⁻] + [HA²⁻] + [A³⁻]
Доли каждой формы рассчитываются по следующим формулам:
α(H₃A) = [H₃A] / C = [H⁺]³ / D
α(H₂A⁻) = [H₂A⁻] / C = Ka₁ * [H⁺]² / D
α(HA²⁻) = [HA⁻] / C = Ka₁ * Ka₂ * [H⁺] / D
α(A³⁻) = [A³⁻] / C = Ka₁ * Ka₂ * Ka₃ / D
Где D (знаменатель) – общий знаменатель для всех долей: D = [H⁺]³ + Ka₁[H⁺]² + Ka₁Ka₂[H⁺] + Ka₁Ka₂Ka₃
Также долю последней формы кислоты можно использовать и в других целях. Например знание α(PO₄³⁻) необходимо для расчёта растворимости Ca₃(PO₄)₂ при разных pH (см. конспект), а также α(Y⁴⁻) для ЭДТА — это диаграмма распределения для 4-основной кислоты (см. конспект).
Важно: Эти формулы выводятся из системы уравнений, включающей константы диссоциации и материальный баланс.
4. Ключевые точки на диаграмме и правила построения
Диаграмма строится в координатах Доля (α) → pH.
Точки пересечения кривых (где доли двух соседних форм равны) являются ключевыми. В этих точках pH равен pKa соответствующей ступени диссоциации.
Когда α(H₃A) = α(H₂A⁻), pH = pKa₁
Когда α(H₂A⁻) = α(HA²⁻), pH = pKa₂
Когда α(HA²⁻) = α(A³⁻), pH = pKa₃
Правило: При pH = pKaₙ концентрации двух соседних форм равны, а доля каждой из них составляет примерно 0.5. Доля остальных форм в этой точке пренебрежимо мала.
5. Алгоритм построения диаграммы (на примере H₃A)
Рассчитайте pKa₁, pKa₂, pKa₃.
Нанесите на ось pH точки, равные pKa₁, pKa₂, pKa₃. Эти точки разделят диаграмму на 4 характерные области.
Определите доминирующую форму в каждой области:
При pH < pKa₁: доминирует полностью протонированная форма H₃A.
При pKa₁ < pH < pKa₂: доминирует промежуточная форма H₂A⁻.
При pKa₂ < pH < pKa₃: доминирует промежуточная форма HA²⁻.
При pH > pKa₃: доминирует полностью депротонированная форма A³⁻.
Рассчитайте доли форм для ряда значений pH (например, с шагом 0.5-1 единица pH) по формулам выше.
Постройте график, соединив рассчитанные точки плавными кривыми для каждой формы.
6. Пример (ортофосфорная кислота H₃PO₄)
pKa₁ ≈ 2.15
pKa₂ ≈ 7.20
pKa₃ ≈ 12.35
Описание диаграммы для H₃PO₄:
При pH < 2.15: в растворе преобладает H₃PO₄.
При pH ≈ 2.15: α(H₃PO₄) = α(H₂PO₄⁻) ≈ 0.5.
При 2.15 < pH < 7.20: доминирует ион H₂PO₄⁻.
При pH ≈ 7.20: α(H₂PO₄⁻) = α(HPO₄²⁻) ≈ 0.5.
При 7.20 < pH < 12.35: доминирует ион HPO₄²⁻.
При pH ≈ 12.35: α(HPO₄²⁻) = α(PO₄³⁻) ≈ 0.5.
При pH > 12.35: доминирует ион PO₄³⁻.
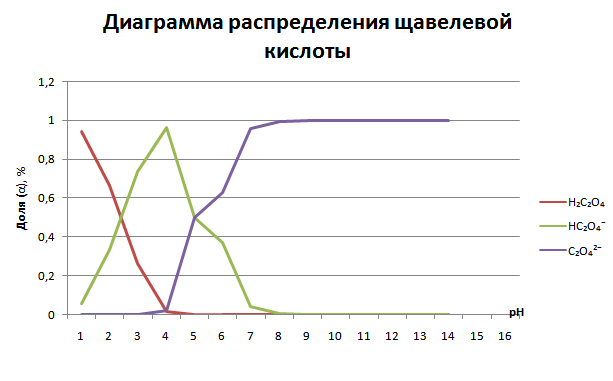
Важно: есть такое понятие, как “хорошо разделенные рКа”, суть заключается в том, что значение рКа двух соседних форм кислоты должны достаточно различаться друг от друга (больше, чем 3), чтобы их можно было эффективно разделить друг от друга и четко видеть переход от одной формы, к другой. У фосфорной кислоты это правило соблюдается и мы четко видим переход из одной области в другую. А вот у щавелевой кислоты разность рКа уже меньше, чем 3 и вторая форма на графике никогда не будет доминирующей.
7. Практическое применение диаграмм распределения
Буферные растворы: Области, где кривые двух форм пересекаются (вокруг pKa), являются зонами буферирования. Наибольшая буферная емкость наблюдается при pH = pKa.
Расчёт равновесных концентраций: Позволяет легко определить, какая форма кислоты преобладает при заданном pH.
Аналитическая химия: Используется для выбора условий титрования, индикаторов, разделения веществ.
Биохимия:Объяснение поведения аминокислот, белков и других биомолекул, которые являются полипротонными.
Вывод: Диаграмма распределения – это мощный визуальный инструмент для анализа кислотно-основного равновесия в растворах полипротонных кислот, связывающий концентрации всех форм с pH раствора и константами диссоциации.
Расчёт растворимости труднорастворимых электролитов с учётом ионной силы и pH
Ученые начала XX века столкнулись с проблемой: классическая теория электролитов переставала работать с увеличением концентрации растворов. Экспериментальные данные всё больше расходились с теоретическими предсказаниями. Нужен был новый подход.
Ионная сила и активность
В 1923 году Питер Дебай и Эрих Хюккель предложили элегантное решение. Они ввели новую величину — ионную силу, которая стала мерой «неидеальности» раствора. Это позволило перейти от концентраций к активностям — эффективным концентрациям, с которыми ионы реально участвуют в процессах.
Обычные концентрации описывают идеальные растворы. Активность же наоборот используются для описания неидеальных растворов.
I=(1/2)*Σ(Zi2 * Ci)
В этой формуле I — это ионная сила, Z — заряд иона, C — концентрация иона
Чем выше значения ионной силы тем выше отклонение от идеальности.
Установлена связь концентрации и активности через коэффициент активности.
a=f*c
Где f — коэффициент активности. Коэффициент активности равный 1 характеризует идеальный раствор. Но в неидеальных растворах он может быть и больше, и меньше 1. Это соответствует положительным и отрицательным отклонениям от идеальности. Обычно при решении задач значения лежат в диапазоне от 0,01 до 10.
Для расчета коэффициента активности был предложен набор формул для разных значений ионной силы:
1)I меньше или равна 0,01М
lg f = 0,509*Z2*(I)½
2)I в интервале от 0,01 до 0,1
lg f = -(0,509*Z2(I)½)/(1+a*B*(I)½)
Здесь a — эмпирическая константа, которая зависит от размера иона, характеризует среднее расстояние на которое сближаются сольватированные ионы.
a=(3-4)*10(-8)
B=0.33*108 эта константа зависит от температуры и диэлектрической проницаемости растворителя
3)I от 0.1 до 0.5 М. В этом случае используется уравнение Девиса.
lg f = -((A*Zi2*I1/2)/(1+aB*I1/2)) + C*I
Всё это необходимо для описания равновесия в растворе, в которых ионная сила не равна 0 и возможно присутствие индифферентного элемента. Это такой элемент, который хоть и присутствует в растворе, но не принимает непосредственного участия в реакции.
Растворимость
Труднорастворимые электролиты образуют систему “осадок — раствор над раствором”, например, хлорид серебра и вода, карбонат кальция и вода, порошок тиомочевины и ацетон. Эта система называется гетерогенной, и она характеризуется двумя характеристиками: растворимость и константа растворимости.
Растворимость — это концентрация вещества в растворе над осадком. Выражается в виде М (моль/л).
На растворимость влияют температура (при нагревании увеличивается) и pH раствора (наиболее важный фактор для рассмотрения, если в состав осадка входят ионы, образующие с частицей H+ слабый электролит)
Для расчета растворимости (S) будет пользоваться константой растворимости (Ks).
Константа растворимости — это величина, которая характеризует растворимость малорастворимых веществ в насыщенном растворе при определенной температуре.
Ks используется для идеальных растворов.
Пример расчета системы без побочных реакций
Пусть у нас есть гетерогенная система: AgCl (осадок) = Ag+ + Cl- (раствор над осадком). Ks для такой системы будет равна: Ks = [Ag+]*[Cl-].
В идеальных условиях равновесная константа равновесия (та, которая была написана выше Ks) равна термодинамической(K0s). Термодинамическая константа используется для описания неидеальных растворов, в которых существуют взаимодействия между молекулами.
K0s = а(Ag+) * a(Cl-)
K0s = [Ag+]*[Cl-]*f(Ag+)*f(Cl-)
K0s = Ks*f(Ag+)*f(Cl-)
Пользуясь этой формулой можно рассчитать растворимость соединения в неидеальном растворе.
Равновесные концентрации, например, [Ag+], складываются из двух величин: стехиометрических коэффициентов и растворимости (S), т.е. [Ag+] = 1*S. Отсюда мы можем находить численное значение растворимости.
Как в случае с растворимостью, так и с устойчивостью комплексов (Хромов «Расчёт растворимости труднорастворимых электролитов с учётом ионной силы и pH») , истинная термодинамическая константа — величина постоянная. Но в реальных условиях её нужно корректировать на доли активных форм, чтобы получить условную константу, применимую к конкретным условиям (pH, ионная сила)
Расчет растворимости труднорастворимых электролитов с учетом ионной силы
Рассчитайте растворимость (S) CaCO3 в водном растворе NaNO3
CaCO3 + H2O + NaNO3
NaNO3 хоть и является индифферентным элементом, но вносит изменения в ионную силу, поэтому ионная сила для этого раствора не равна нулю. Следовательно, пользуемся формулой, выведенной ранее.
Растворимость CaCO3 в идеальном растворе, т.е. в растворе с ионной силой равной нулю, равна 6,2*10-5 моль/л. Значит, что при введении индифферентного NaNO3 значение растворимости выросло.
Расчет растворимости труднорастворимых электролитов с учетом pH раствора
Следующий фактор, который может влиять на растворимость – это pH раствора.
Пусть у нас есть тот же водный раствор CaCO3, но его pH = 6. Что в таком случае будет с растворимостью?
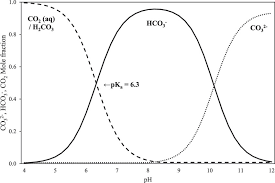
Так как угольная кислота может существовать в нескольких формах в зависимости от pH, то мы должны учитывать каждую возможную форму в растворе.
В данном случае используется условная константа растворимости, т.к. в растворе протекают побочные процессы, которыми мы не в силе пренебречь. В данном случае из-за pH образуется слабая угольная кислота, которая после разлагается на H2O и CO2.
Таким образом, для понимания поведения труднорастворимых электролитов в растворе в реальных системах необходимо учитывать ионную силы, pH раствора, возможные побочные реакции. Изученные формулы и термины являются неотъемлемой частью отхождения от идеальных моделей к неидеальным.
Анализ изотермы адсорбции по БЭТ: определение удельной поверхности твёрдого тела.
1. Введение
Удельная поверхность (Sуд) — это площадь поверхности твёрдого тела, отнесённая к его единице массы (м²/г). Чем мельче частицы и чем больше в материале пор, тем выше этот параметр. Его знание критически важно для создания эффективных катализаторов, сорбентов, фильтров и аккумуляторов.
2. Адсорбция: основные понятия
Адсорбция — это увеличение концентрации вещества на границе раздела фаз (например, газа на поверхности твёрдого тела). Поглощение веществом по всему объёму — это абсорбция.
Адсорбент — твёрдое тело, на поверхности которого идёт адсорбция (например, активированный уголь).
Адсорбтив — вещество, которое адсорбируется (например, газообразный азот).
3. Изотерма адсорбции
Изотерма адсорбции — это график зависимости количества адсорбированного газа от его давления (или относительного давления P/P₀) при постоянной температуре. Для пористых материалов чаще всего получают изотермы II и IV типов по IUPAC.
4. Теория БЭТ
Визуальная схема модели БЭТ (Брунауэра-Эммета-Теллера) иллюстрирует многослойную физическую адсорбцию на поверхности твердого тела. Графическое пояснение включает в себя изображение адсорбента с пористой поверхностью и молекул адсорбата (например, газа), которые образуют один слой на поверхности, а затем последующие слои над первым, при конденсации газа на уже адсорбированных молекулах.
Основные положения модели БЭТ
Многослойная адсорбция: В отличие от модели Ленгмюра, которая описывает монослойную адсорбцию, модель БЭТ учитывает образование нескольких слоев адсорбированных молекул.
Предположения:
Поверхность адсорбента считается однородной.
Взаимодействие между адсорбентом и адсорбатом является более сильным, чем взаимодействие между молекулами адсорбата.
Взаимодействие между молекулами адсорбата учитывается только в направлении, перпендикулярном поверхности, и рассматривается как процесс конденсации.
Графическое пояснение
На схеме модель БЭТ изображается как пористая структура адсорбента.
Первый слой: Молекулы газа адсорбируются на поверхности адсорбента. Это соответствует монослою адсорбата, который показан непосредственно на поверхности пор.
Последующие слои: Когда адсорбированный слой полностью сформирован, дальнейшие молекулы газа продолжают адсорбироваться на поверхности первого слоя, образуя второй слой. Этот процесс продолжается, пока не будет адсорбировано несколько слоев.
Учет взаимодействия: Схема показывает, что адсорбированные молекулы в последующих слоях взаимодействуют как молекулы в конденсированном газе. Такое взаимодействие происходит в направлении, перпендикулярном поверхности.
Модель в целом: Визуализация модели БЭТ помогает понять, как адсорбция происходит в многослойном режиме и как можно рассчитать площадь поверхности материала, исходя из объема адсорбированного газа.
Примечание: Модель БЭТ широко используется для оценки удельной площади поверхности пористых материалов, например, при анализе адсорбции азота.
Метод БЭТ обобщает модель Ленгмюра (см. Симовин) на случай нескольких слоёв. При P/P₀ → 0 изотерма БЭТ переходит в изотерму Ленгмюра
5. Практическое применение метода БЭТ
Уравнение БЭТ в линеаризованной форме — главный инструмент расчёта:
P/P0 — относительное давление газа
a — количество адсорбированного вещества при данном относительном давлении газа
am — искомая мономолекулярная ёмкость
C — константа, связанная с энергией адсорбции.
Как работать с уравнением:
Проводят эксперимент по адсорбции азота при температуре кипения 77К
Строят график в координатах P/P0 по оси X, а [P/P0]/[a(1-P/P0)] по оси Y
Находят линейный участок в диапазоне P/P00.05-0.35
По углу наклона (k) и отрезку, отсекамому на оси Y(b), рассчитывают:
am = 1/(k+b)
С=k/b + 1
На практике построение графиков БЭТ сегодня почти всегда делается в автоматизированных системах (например, NovaWin, Micromeritics, BET Surface Area Calculator и др.).
6. Расчет удельной поверхности.
Зная am (объём газа, необходимый для покрытия одного монослоя), переходят к удельной поверхности
σ — площадь поперечного сечения одной молекулы адсорбата (для N2 0,162 нм2)
m — масса образца адсорбента
Vm — молярный объём газа
Примечание:
Значение σ = 0.162 нм² для N₂ при 77 К принято в большинстве стандартов (ISO, IUPAC), но в научной литературе могут встречаться значения от 0.14 до 0.18 нм². Это — основной источник систематической погрешности в методе БЭТ
Дано:
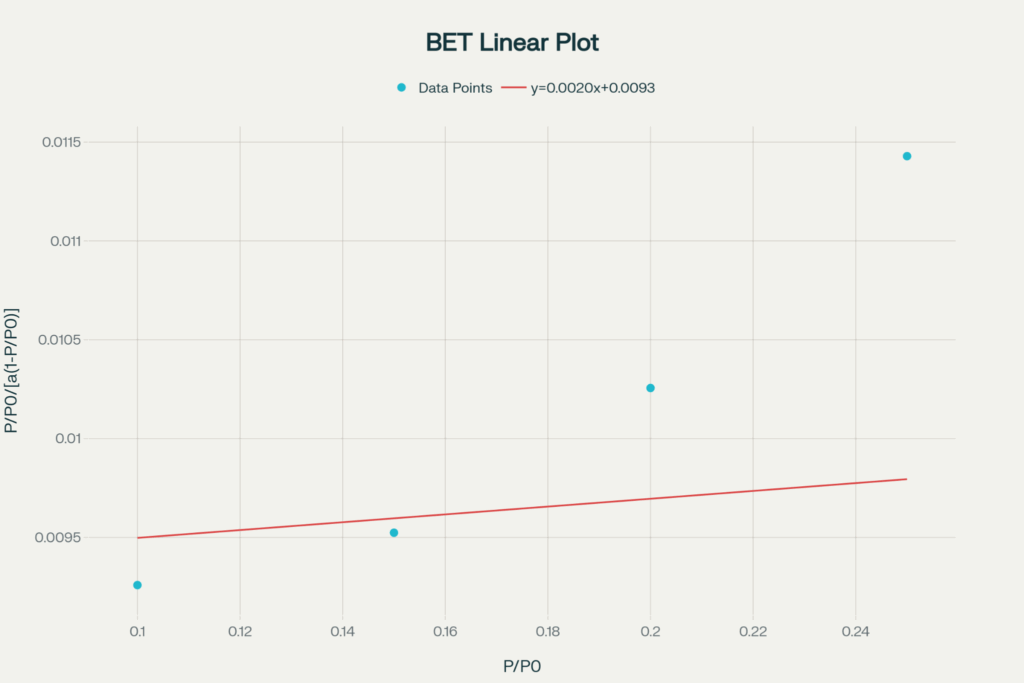
При P/P₀ = 0.10, 0.15, 0.20, 0.25
a = 12, 18, 25, 35 см³/г
Построить график БЭТ → найти k и b → вычислить aₘ → рассчитать Sуд.
По формуле БЭТ рассчитываем для каждого значения
y=P/P0/(a(1-P/P0))
Строим график зависимости y от x=P/P0. Линейная аппроксимация даёт уравнение прямой: y=kx+b
Коэффциенты:
k = 0.00198
b = 0.00930
Дальше по формуле рассчитываем объём монословая am:
am=1/(k+b)=88,65 см3/г
Удельную поверхность рассчитываем по формуле:
Sуд=amNAs/Vm
Где число Авогадро NA=6.022*1023, площадь одной молекулы азота s=0,162*10-18 м2. Молярный объём Vm=22400 см3/моль, что даёт:
Sуд=386.1 м2/г.
Ниже приведён график БЭТ с экспериментальными точками и аппроксимирующей прямой линии.
7. Ограничения метода БЭТ
Метод БЭТ даёт хорошие результаты для материалов с развитой поверхностью (Sуд > 1–5 м²/г). Он менее точен для непористых или микропористых материалов, где преобладает мономолекулярная адсорбция и модель полимолекулярного покрытия не работает.
Вступающие в химическую реакцию вещества и ее продукты обладают разным запасом энергии. Это приводит к тому, что в ходе реакции должна выделяться теплота, если суммарная энергия продуктов меньше чем у реагентов, и, напротив, требуется дополнительная энергия из окружающей среды на образование продуктов, которые должны обладать более высоким запасом энергии по сравнению с исходными. Эти тепловые характеристики связаны с прочностью (энергией) химических связей
Таким образом, любая химическая реакция будет обладать тепловым эффектом:
Тепловой эффект — энергия (теплота), которая выделяется или поглощается в ходе протекания химической реакции. Она измеряется в килоджоулях (кДж)
Расчеты с участием теплового эффекта находят свое применение в самых различных областях химии. К примеру, они позволяют анализировать поведение веществ в разнообразных реакциях, сравнивая их энергетические параметры, а также подбирать оптимальные условия для протекания химических процессов, что имеет важное значение для достижения максимальной эффективности на промышленных установках.
Данные о теплотах реакций имеют большое значение при изучении химической кинетики, т.к. тепловые характеристики веществ сильно влияют на скорость и направление химических реакций.
Также тепловые эффекты могут использоваться в аналитической химии для качественного и количественного анализа
Классификация реакций по тепловому эффекту
Если в ходе реакции теплота выделяется (знак теплового эффекта «+»), то такая реакция является экзотермической: 2NO + O2 → 2NO2 + 114 кДж.
При окислении двух молей NO по этой реакции выделяется 114 кДж. Это связано с образованием оксида NO2 с более прочными химическими связями — их формирование энергетически выгодно, поэтому излишняя энергия выделяется в окружающую среду в виде теплоты
Если в ходе реакции теплота поглощается (знак теплового эффекта «-»), то такая реакция является эндотермической:
N2 + O2 → 2NO – 180,8 кДж
При окислении одного моля N2 поглощается 180.8 кДж. Молекула азота является крайне стабильной. Валентные связи в ней крайне прочные и для их разрыва требуются большие затраты энергии из внешней среды. Именно поэтому азот, составляющий 78% земной атмосферы, не вступает самопроизвольно во многие потенциальные химические реакции, оставаясь достаточно инертным веществом
Такие уравнения реакций, для которых указано количество выделяющейся или поглощаемой в ее ходе теплоты, называются термохимическими уравнениями
С термохимическими уравнениями можно работать как с математическими выражениями, т.е. их можно складывать, вычитать или же умножать на какое-то число. Т.к. часто тепловой эффект реакции рассматривается только для одного моля вещества, то стехиометрические коэффициенты, стоящие при других участниках реакции, могут оказываться дробными величинами. Записывать термохимические уравнения с дробными коэффициентами возможно, т.к.тепловой эффект указывается в расчете на моль вещества, т.е. эти уравнения оперируют количеством вещества, а не молекулами
СН4 +3/2О2 = СО + 2Н2О(Г) + 519,33 кДж
Правило для задач: Как видно из этих уравнений тепловой эффект всегда пропорционален количеству одного из веществ (любого), участвующего в химической реакции. Это правило используется в расчетных задачах
Влияние изменения температуры на смещение химического равновесия
В соответствие с принципом Ле Шателье можно сказать, что:
При повышении температуры химическое равновесие системы смещается в сторону эндотермической реакции, а при понижении температуры – в сторону экзотермического процесса
H2 (г) + Cl2 (г) ⇄ 2HCl (г) + Q
Реакция является экзотермической, т.е. идёт с выделением теплоты, поэтому при нагревании ее равновесие сместится влево, в сторону исходных веществ, а при уменьшении температуры — наоборот, вправо, в сторону образования продуктов.
C4H10 (г) ⇄ C4H8 (г) +H2 (г) — Q
Реакция является эндотермической — она идет с поглощением теплоты извне, поэтому при нагревании ее равновесие сместится в сторону образования продуктов, а при уменьшении температуры — в сторону исходных веществ.
Какие выводы можно сделать из ТХУ?
является ли реакция эндотермической или экзотермической
позволяет проанализировать устойчивость соединений и прочность образующихся связей
позволяет подобрать более оптимальные условия для протекания реакции
рассчитать, какое количество вещества вступило в реакцию и какое количество продукта образовалось, если известен тепловой эффект реакции и наоборот
Существуют некоторые закономерности, которые помогают в некоторых случаях предсказать знак теплового эффекта.
Например, реакции, которые протекают самопроизвольно при нормальных условиях, чаще всего являются экзотермическими. В качестве примера можно привести горение угля. Эта реакция протекает самопроизвольно и необратимо с выделением большого количества тепла, т.е. реакция является экзотермической
Типичными экзотермическими реакциями являются:
реакции горения
реакции активных металлов и их оксидов с водой или с кислотами
реакции нейтрализации между щелочами и сильными кислотами
большинство легко протекающих реакций соединения (исключения: взаимодействие азота с кислородом и некоторые другие реакции)
Типичными эндотермическими реакциями являются:
реакции разложения
фотосинтез
Дополнительные термины
Отдельно выделяют теплоту образования и сгорания вещества, которые используют при описании химического соединения:
Теплота образования вещества – количество теплоты, выделяющееся при образовании 1 моль данного вещества из простых веществ.
Например, при сгорании алюминия:
2Аl + 3/2О2 → Аl2О3 + 1675 кДж
теплота образования оксида алюминия равна 1675 кДж/моль.
Если мы запишем термохимическое уравнение без дробных коэффициентов:
4Аl + 3О2 → 2Аl2О3 + 3350 кДж
теплота образования Al2O3 все равно будет равна 1675 кДж/моль, т.к. в термохимическом уравнении приведен тепловой эффект образования 2 моль оксида алюминия.
Теплота сгораниявещества – количество теплоты, выделяющееся при горении 1 моль данного вещества.
Например, при горении метана:
СН4 + 2О2 → СО2 + 2Н2О + 802 кДж
теплота сгорания метана равна 802 кДж/моль.
Иногда в некоторых физико-химических задачах требуется вычислить теплоту, которую нужно затратить на нагревание веществ до определенной температуры или для осуществления фазового перехода (например, когда меняется агрегатное состояние воды: лед-жидкость-пар). В этом случае пользуются новыми термодинамическими величинами
Удельная теплоемкость вещества — это физическая величина, которая показывает, какое количество теплоты в Дж необходимо передать веществу массой 1 кг для его нагрева на 1°C
Значения удельной теплоемкости различных веществ приводятся в справочниках. Так для воды (ж.) удельная теплоемкость равна 4200 Дж/кг*1°C
Задачи на тепловой эффект
Пример 1 задачи на расчеты с тепловым эффектом
Дано термохимическое уравнение для реакции сгорания водорода:
2H2(г) + O2(г) = 2H2O(г) + 484 кДж
Необходимо найти массу образовавшейся воды в граммах, если известно, что в ходе реакции выделилось 1479 кДж теплоты
Решение:
Из термохимического уравнения видно, что при образовании двух моль воды выделяется (т.к. реакция экзотермическая) 484 кДж теплоты. Вычислить массу, выделившейся в ходе реакции воды можно, составив простую пропорцию:
2 моль H2O — 484 кДж x моль H2O — 1479 кДж
m(H2O) = n(H2O)*M(H2O) = 6.1 моль*18 г/моль = 110 г
Пример 2 задачи на расчеты с тепловым эффектом
В ходе реакции сгорания 8.96 л этана при нормальных условиях выделилось 588 кДж теплоты. Определите теплоту сгорания этана в этих условиях.
Теплота сгорания — это количество теплоты, которые выделится при сгорании одного моль вещества. В соответствии с этим определением запишем уравнение реакции для одного моль этана C2H6. Мы воспользуемся дробными коэффициентами:
Рассчитаем количество этана в молях: n(C2H6)=8.96/22.4 = 0.4 моль
Тогда для решения задачи достаточно составить простую пропорцию:
0.4 моль C6H6 — 588 кДж 1 моль C6H6 — x кДж
Пример 3 задачи на расчеты с тепловым эффектом
Дано термохимическое уравнение сгорания метана
Рассчитайте количество литров (н.у) метана, которое понадобится для того, чтобы вскипятить 2л воды, которая изначально имеет температуру 20°C. Удельная теплоемкость воды равна 4200 Дж/кг*°C
Для начала найдем количество теплоты, которое нужно затратить для того, чтобы довести воду до точки кипения
Теперь составим пропорцию
1 моль CH4 — 891 кДж x моль CH4 — 672 кДж
В этом случае понадобится 0.75*22.4 = 16.8 л CH4
Проверить свои знания по теме «Термохимия» можно в этом тестировании:
Химическое равновесие-состояние химической системы, когда скорость прямой реакции равна скорости обратной реакции. Для системы, находящейся в состоянии равновесия, концентрация реагентов, температура и другие параметры системы постоянны.
При решении задач на реакторы удобно воспользоваться таблицей, в которую мы записываем реагенты, изменение концентрации веществ в ходе реакции и равновесные, исходные концентрации. То, что нужно найти, можно обозначить как “X” и “Y”. Исходная концентрация вещества, полученного в ходе реакции, равна нулю. В ходе реакции концентрации реагентов уменьшаются, а концентрации продуктов увеличиваются. Если в условии задачи сказано, что объем постоянен, то для реагента концентрация и его количество вещества равны.
Рассмотрим пример задачи на реакторы:
В реактор постоянного объёма ввели бутан и сильно нагрели. В реакторе установилось равновесие:
При этом исходная концентрация бутана составила 8 моль/л, а равновесная концентрация этилена — 6 моль/л.
Определите равновесные концентрации и
Составим таблицу:
В первой строке запишем реагенты, во второй — исходные концентрации веществ, в третьей — изменение концентраций веществ, в четвёртой — равновесные концентрации.
Реагенты
Исходные конц.
Изменение конц.
Равновесные конц.
В первую строку записываем реагенты с учетом коэффициентов, стоящих перед ними. Так, мы получаем 1 C4H10, 2 C2H4 и 1 H2.
Реагенты
1C4H10
2C2H4
1H2
Исходные конц.
Изменение конц.
Равновесные конц.
Затем заполняем известные нам данные — исходную концентрацию бутана и равновесную концентрацию этилена, равные 8 моль/л и 6 моль/л соответственно. Обозначим за “X” равновесную концентрацию бутана, за “Y” равновесную концентрацию водорода и зафиксируем их в таблице.
Реагенты
1C4H10
2C2H4
1H2
Исходные конц.
8 моль/л
Изменение конц.
Равновесные конц.
X
6 моль/л
Y
Далее переходим к заполнению третьей строки. Начинаем заполнение с того вещества, для которого даны и равновесная, и исходная концентрации; в нашем случае это этилен. Исходная концентрация этилена равна нулю, так как изначально он не присутствовал в реакционной системе,а его равновесная концентрация равна 6 моль/л, следовательно, концентрация этилена увеличилась на 6 моль/л. Записываем в третью строку под этиленом “+6” (“+” указывает на увеличение концентрации).
Реагенты
1C4H10
2C2H4
1H2
Исходные конц.
8 моль/л
0 моль/л
0 моль/л
Изменение конц.
+6
Равновесные конц.
X
6 моль/л
Y
Исходя из реакции мы видим, что на образование 2 моль этилена и 1 моль водорода расходуется 1 моль бутана. Отсюда следует, что на образование 6 моль этилена затратилось 3 моль бутана, а количество образовавшегося в результате реакции водорода в 2 раза меньше, чем количество образовавшегося этилена (3 моль), так как перед этиленом стоит коэффициент 2, а перед водородом 1. Запишем в третью строку под водородом +3, а под бутаном -3(из-за того, что бутан расходуется в ходе реакции, а водород образуется).
Реагенты
1C4H10
2C2H4
1H2
Исходные конц.
8 моль/л
0 моль/л
0 моль/л
Изменение конц.
-3
+6
+3
Равновесные конц.
X
6 моль/л
Y
Как мы видим, исходная концентрация водорода равна нулю, а в ходе реакции она увеличилась на 3 моль/л, следовательно, равновесная концентрация водорода равна: 0+3=3 моль/л (равновесная концентрация равна сумме или разности между исходной концентрацией и изменением концентрации в ходе реакции). Так как в ходе реакции концентрация бутана уменьшилась на 3 моль/л, а его исходная концентрация равна 8 моль/л, то 8-3=5 моль/л — равновесная концентрация бутана.
Рассмотрим следующую задачу:
В реактор постоянного объёма ввели азотный ангидрид и повысили температуру. В реакторе установилось равновесие:
При этом равновесные концентрации оксида азота(V) и оксида азота(IV) составили 0,02 моль/л и 0,16 моль/л соответственно.
Определите исходную концентрацию и равновесную концентрацию
Заполняем таблицу по тому же принципу, что и в прошлой задаче.
Реагенты
2N2O5
4NO2
O2
Исходные конц.
X
0 моль/л
0 моль/л
Изменение конц.
Равновесные конц.
0,02 моль/л
0,16 моль/л
Y
Отличие этой задачи от прошлой заключается в том, что сейчас нам нужно найти исходную и равновесную концентрации веществ, а ход решения один и тот же. На образование 4 моль NO2 и 1 моля O2 было затрачено 2 моля N2O5, следовательно, на образование 0,16 моль NO2 было израсходовано 0,08 моль N2O5, а количество вещества кислорода в 4 раза меньше, чем оксида азота (IV), и равно 0,04 моль.
Реагенты
2N2O5
4NO2
O2
Исходные конц.
X
0 моль/л
0 моль/л
Изменение конц.
-0,08 моль/л
+0,04 моль/л
Равновесные конц.
0,02 моль/л
0,16 моль/л
Y
Равновесная концентрация кислорода равна 0+0,04=0,04 моль/л. Исходная концентрация N2O5 больше его равновесной на то количество, которое было израсходовано в ходе реакции, т.е. исходная концентрация оксида азота (V) равна: 0,02+0,08=0,1 моль/л.
Еще один пример задачи на реакторы:
В реакторе постоянного объема смешали этилен и пары воды в мольном соотношении 1:2. Смесь нагрели и добавили катализатор. Через некоторое время установилось равновесие:
При этом исходная концентрация этилена составила 0,3 моль/л, а равновесная концентрация воды — 0,35 моль/л. Найдите исходную концентрацию и равновесную концентрацию
Составим таблицу, аналогичную таблицам из прошлых задач.
Реагенты
1C2H4
1H2O
1C2H5OH
Исходные конц.
0,3 моль/л
X
0 моль/л
Изменение конц.
Равновесные конц.
0,35 моль/л
Y
Так как объем системы постоянен, то количества веществ в системе пропорциональны их концентрациям. Согласно уравнению обратимой реакции на образование 1 моль этанола тратится 1 моль этилена и 1 моль воды. В условии сказано, что смесь этилена и воды была в мольном соотношении 1:2, значит, исходная концентрация воды в два раза больше концентрации этилена и равна 0,6 моль/л.
Реагенты
1C2H4
1H2O
1C2H5OH
Исходные конц.
0,3 моль/л
X=0,6 моль/л
0 моль/л
Изменение конц.
-0,25 моль/л
+0,25 моль/л
Равновесные конц.
0,35 моль/л
Y
В реакцию вступило 0,6-0,35=0,25 моль/л воды. Равновесная концентрация этанола равна концентрации вступившей воды в реакцию, т. е. 0,25 моль/л.
Химическое равновесие. Факторы, влияющие на смещение химического равновесия.
Химические реакции могут быть обратимыми и необратимыми.
Необратимыми называются те реакции, которые идут только в одном (прямом) направлении.
Обратимыми же называют те реакции, которые идут как в прямом, так и в обратном направлении.
Рассмотрим пример.
Пусть протекает реакция:
А + В = С + D
В том случае, если данная реакция необратима, обратный процесс, т.е. взаимодействие С и D с образованием A и B не протекает, а имеет место лишь взаимодействие A и В. Если же, наоборот, данная реакция обратима, то, значит, что имеет место как прямой, так и обратный процесс, т.е. реакция идёт как в прямом, так и в обратном направлении.
Примеры обратимых реакций:
N2 + 3H2 = 2NH3
Н2O + СO2 = Н2СO3
СН3СООН + С2Н5ОН = СН3СООС2Н5 + Н2О
Примеры необратимых реакций:
NaOH + HCl = NaCl + H2O
Ca(NO3)2 + 2KF = CaF2 + 2KNO3
Когда только запускается обратимая реакция, то скорости прямого и обратного процесса неодинаковы. Со временем же протекания реакции взаимодействия, в какой-то момент, скорости прямой и обратной реакций выравниваются между собой. И наступает состояние химического равновесия.
Итак, если система находится в состоянии химического равновесия, то скорости прямой и обратной реакций равны между собой. Это в свою очередь и обеспечивает постоянство концентраций реагентов и продуктов реакции. Посмотрим на примерах, что будет происходить с системой, если на неё оказать влияние извне. Оказание воздействие на систему может происходить за счёт следующих факторов:
Изменение концентрации исходных веществ и продуктов реакции
Изменение температуры
Изменение давления (только для реакций с участием газов)
Для того, чтобы определять, каким образом сдвигается химическое равновесие при том или ином воздействии, пользуются принципом Ле Шателье.
«Если на уравновешенную систему действовать извне, то система переходит в такое состояние, в котором эффект внешнего воздействия ослабевает»
С соответствие в данным принципом мы можем определить направление смещения химического равновесия. Переход системы из одного равновесного состояния в другое равновесное состояние называют сдвигом химического равновесия.
Когда система находится в состоянии химического равновесия, то скорости прямой и обратной реакций равны. При изменении же указанных выше факторов, какая-то из скоростей (или прямой или обратной реакции) будет иметь большее значение по сравнению с другой. В этом и кроется причина смещения(сдвига) химического равновесия.
Влияние изменения концентрации исходных веществ и продуктов реакции на направление смещения равновесия.
В соответствие с принципом Ле Шателье, можно сказать, что:
При увеличении концентрации реагирующих веществ химическое равновесие системы смещается в сторону образования продуктов реакции, а при увеличении концентрации продуктов реакции химическое равновесие системы смещается в сторону образования исходных веществ.
Важно отметить, то твердые вещества не оказывают влияния на состояние химического равновесия. То есть изменение их количества в системе не приводит ни к какому эффекту!
Рассмотрим реакции:
Заполним таблицы, которые относятся к данным реакциям, в соответствие с вышеописанным правилом. Стрелками будем показывать направление смещения химического равновесия.
2NO(г) + O2(г) → 2NO2(г)
N2O4(г) ⇄ 2NO2 (г)
Fe(тв) + 4Н2О(г) ⇄ Fe3О4(тв) + 4Н2(г)
Влияние изменения температуры на направление смещения химического равновесия.
Выделяют экзотермические и эндотермические химические реакции. Экзотермические реакции протекают с выделением тепла в окружающую среду, а эндотермические протекают с поглощением тепла из окружающей среды. То есть эти реакции отличаются по тепловому эффекту.
В соответствие с принципом Ле Шателье можно сказать, что:
При повышении температуры химическое равновесие системы смещается в сторону эндотермической реакции, а при понижении температуры – в сторону экзотермического процесса
Рассмотрим реакции и аналогично заполним таблицы:
H2 (г) + Cl2 (г) ⇄ 2HCl (г)+ Q
C4H10(г) ⇄ C4H8(г) +H2(г)-Q
Влияние изменения давления на направление смещения химического равновесия.
Данный фактор оказывает влияние на смещение химического равновесия только в случае, когда в реакции присутствует газообразное вещество.
При увеличении давления химическое равновесие системы смещается в сторону той реакции, при которой объем образующихся газообразных веществ меньше, а при уменьшении давления– в сторону той реакции, при которой объем образующихся газообразных веществ больше.
Рассмотрим примеры:
CO2(г)+H2(г) ⇄CO(г)+H2O(г)
SO2 (г) + H2O (ж) ⇄ H2SO3 (ж)
C2H6(г)⇄C2H2(г)+ 2H2(г)
Можно также сказать, что на смещение равновесия влияет изменение объема реакционного сосуда. Это утверждение аналогично справедливо только для газов. Чем меньше давление газа, тем больший объем он занимает и наоборот. Поэтому рассмотрение процесса смещения химического равновесия относительно объёмов можно легко перевести на давление. Эти понятия связаны, так как с увеличением давления происходит уменьшение объёма, а с уменьшением давления – увеличение объема.
Скорости химических реакций ускоряются с помощью катализаторов. Но поскольку катализатор в равной степени ускоряет как прямую, так и обратную реакции, то его наличие не оказывает влияние на состояние химического равновесия!
Состояние химического равновесия – это состояние, при котором имеют место химические реакции и, значит, система имеет определенную динамику. Смещение же химического равновесия всегда приводит к тому, что в ходе него устанавливается новое состояние равновесия, с новыми концентрациями исходных веществ и продуктов реакции.
Термодинамика как наука появилась в середине 19 столетия. Тогда целью термодинамики было установление взаимосвязи между работой и теплотой, а также разработка теории паровой машины. В дальнейшем цели термодинамики значительно расширяются и все ее основные законы и положения стали широко применяются в различных областях науки особенно в химии.
Термодинамика – раздел физики, изучающий способы передачи энергии, путём взаимного превращения теплоты и работы.
В классической термодинамике изучаются свойства макроскопических систем в целом, а отдельные частицы не рассматриваются.
Макроскопическая система – это система, состоящая из большого количества частиц (молекул, атомов, ионов), поведение которых можно описать законами статистики и теории вероятности.
Термодинамика базируется на трех законах, из которых путем логических умозаключений можно получить остальные положения данной науки. Как было сказано ранее термодинамика базируется на двух постулатах и трёх законах (началах), которые не выводятся теоретически, а представляют собой следствия, обобщающие опыт человека.
В данном конспекте мы рассмотрим лишь первый закон термодинамики, его применение, а также некоторую часть других положений.
Для понимания последующего материала рассмотрим ряд наиболее основных понятий и терминов.
Системы
Под термодинамическойсистемой тело или группа тел, мысленно выделяемые из окружающей среды.
Термодинамические системы подразделяются на:
гомогенные системы, в которых все физические и химические свойства во всех частях системы одинаковы. Например, чистая вода или раствор поваренной соли в воде, воздух.
гетерогенные системы, состоящие из разнородных тел, отделенных друг от друга видимыми поверхностями раздела. Примерами гетерогенных систем служат: насыщенный раствор с осадком, смесь воды и бензина, вода с песком.
Фаза – гомогенная часть гетерогенной системы.
Виды систем
Изолированная система – система, которая не обменивается с окружающей средой ни веществом, ни энергией. Такая система характеризуется постоянством внутренней энергией и объема (U=const, V=const).
Закрытая система – не обменивается веществом с окружающей средой, но энергообмен возможен (V=const).
Открытая система, способная обмениваться веществом и энергией с внешней средой.
Процессы
Самопроизвольные – происходят без затраты энергии;
Несамопроизвольные – происходят только при затрате энергии;
Равновесными (квазистатическими) называютсяпроцессы, в которых система под действием бесконечно малых воздействий переходит из одного равновесного состояния в другое настолько медленно, что все промежуточные состояния можно рассматривать как равновесные. Равновесные процессы протекают бесконечно медленно, поэтому их ещё называют квазистатическими процессами.
Обратимые – когда переход системы из одного состояния в другое происходит через последовательность одних и тех же состояний, и после возвращения в исходное состояние в окружающей среде не остается никаких изменений;
Необратимые – процессы, в результате которых невозможно возвратить систему и всё её окружение в исходное состояние.
Параметры
Термодинамические параметры – величины, характеризующие термодинамическое состояние системы и выражающие свойства больших групп молекул.
Исходные положения термодинамики
Первый из постулатов, лежащих в основе термодинамики говорит:
Любая изолированная система с течением времени переходит в равновесное состояние и не может самопроизвольно из нее выйти.
Второй постулат термодинамики (закон термического равновесия):
Всякая равновесная система характеризуется температурой — физической величиной, описывающей внутреннее состояние этой системы. Если каждая из двух термодинамических систем находятся в тепловом равновесии с третьей, то они находятся в тепловом равновесии с друг с другом.
Внутренняя энергия. Работа и теплота
С точки зрения молекулярно-кинетической теории внутренняя энергия – это суммарная энергия макроскопического тела. А именно сумма кинетических энергий беспорядочного движения всех частиц тела и потенциальных энергий взаимодействия всех частиц друг с другом.
U = Uпост + Uвращ + Uколеб + Uядер + Uе̄̄
Внутренняя энергия зависит от природы вещества, температуры и количества вещества.
Вычислить абсолютное значение внутренней энергии невозможно, поскольку невозможно учесть движение всех частиц и их взаимное положение из-за огромного числа молекул в макроскопических телах. Но можно рассчитать её изменение их первого закона термодинамики, определяя тепловой эффект и совершенную работу.
Внутренняя энергия является функцией состояния. То есть ее изменение не зависит от пути перехода из состояния 1 в состояние 2.
∆UI = ∆UII = ∆UIII
Теплота – это форма передачи энергии путем излучения или теплопроводности, т.е. путем хаотических столкновений молекул двух соприкасающихся тел.
Работа – передача энергии путем перемещения массы или заряда под действием каких-либо внешних сил, либо против внешних сил. Например, расширение или сжатие газа, поднятие тела в поле тяжести Земли, электрическая работа и др.
Работу, совершаемую системой или внешними силами над системой, будем обозначать W.
Легко показать, что совершаемая системой работа W при расширении или сжатии газа равна:
W = p∆V
где p – давление газа; ∆V = V2 – V 1 – изменение объема газа.
Первый закон термодинамики
Первый закон термодинамики непосредственно связан, а точнее является частным случаем закона сохранения энергии, распространенный на тепловые явления. Он позволяет рассчитать тепловые равновесия различных процессов, в том числе и химических реакций, что для нас химиков особенно интересно.
В 1844 – 1854 гг. Д. Джоуль провел опыт, в результате которого установил эквивалентность теплоты и механической работы в циклических процессах.
Результат опыта Джоуля можно записать так:
∮ δQ = ∮ δW (1)
Qцикл = Wцикл (2)
Если рассмотреть нециклический процесс (например, половину опыта Джоуля):
δQ ≠ δW (4)
Обозначив разность элементарной теплоты и работы как dU, получим запись первого закона термодинамики в виде бесконечно малых изменений величин:
dU = δQ — δW(5)
Проинтегрировав выражение (5), для нециклического процесса получим:
∆U = Q -W (7)
Выражение (7) — это запись первого закона термодинамики в интегральной форме.
Формулировки Первого закона
Существует множество формулировок первого закона. Вот несколько из них:
Невозможно создать машину, которая совершала бы работу без затрата эквивалентного количества другого вида энергии. Или что тоже самое вечный двигатель первого рода невозможен.
Энергия не создается и не разрушается, при всех процессах суммарная энергия изолированной системы остаётся постоянной.
Количество теплоты сообщенное системе идет на изменение внутренней энергии системы и на совершение работы газом против внешних сил.
Применение первого закона термодинамики к различным процессам
С помощью первого закона термодинамики можно делать важные заключения о характере протекающих процессов.
Процессы, в которых участвует система, могут протекать при различных условиях. Рассмотрим такие процессы, в которых система представляет собой идеальный газ.
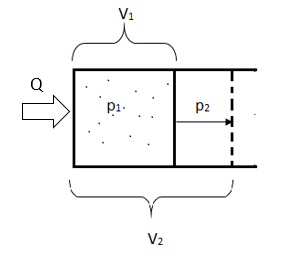
1. Расширение идеального газа в пустоте (вакуум)
Вначале газ находится в сосуде с перегородкой, в одной части которого находится вакуум, а в другой идеальный газ. Когда мы убираем перегородку, газ будет расширяться и в конечном итоге займёт всё пространство сосуда.
В этом процессе изменяется как объем, так и давление. При этом объем увеличивается, а давление уменьшается.
Опыт показывает, что Q=0, работа не совершается, а температура системы не меняется.
Работа равна нулю, так как газ расширяется в вакуум, а, следовательно, внешних сил нет.
Так для идеального газа справедливо:
Таким образом, внутренняя энергия идеального газа зависит только от температуры и не зависит от объема и давления. Это связано с отсутствием межмолекулярного взаимодействия.
2. Изохорное нагревание
При изохорном процессе объем системы остается постоянным.
В данном процессе газ находится под недвижимым поршнем. И если газу подводить теплоту Q газ начнет нагреваться, а давление в сосуде увеличивается.
V=const
dU = δQ — δW
dU = δQ — pdV
Поскольку объём не изменяется, работа не совершается, и получаем, что
dU = δQ
∆U = Qv
В этом процессе вся теплота расходуется на повышение внутренней энергии.
где сV – теплоёмкость вещества при постоянном объеме.
Для данного процесса справедливо выражение:
3. Изотермическое расширение
В данном опыте в некотором цилиндре, закрытом поршнем, способным двигаться без трения, находится идеального газ. Если к газу подводить тепло Q, то он будет расширяться от V1 до V2.
Передача теплоты при T=const от одного тела к другому является квазистатическим процессом. Для идеального газа (а также и для реального при небольших давлениях) внутренняя энергия зависит только от температуры. Отсюда при изотермическом процессе U=const, а изменение внутренней энергии равно нулю.
dU = δQ — δW
δQ = δW
QT = W
Используя уравнение Менделеева-Клапейрона:
pV = nRT
Получаем:
В этом процессе вся подведенная теплота идет на совершение работы расширения.
Для данного процесса справедливо выражение:
4. Изобарное нагревание
К газу, находящемуся под постоянным давлением, подводится количество теплоты Q. Газ в начале находящийся при температуре T1 занимающий объём V1 расширяется до V2и нагревается до T2 при этом давление остаётся постоянным.
p=const
dU = δQ — δW
δQ = dU + pdV
QT = ∆U + p∆V
В этом процессе вся подведенная теплота расходуется как на нагревание, так и на расширение.
где cp — теплоёмкость вещества при постоянном давлении; H – энтальпия.
Следует отметить, что для всех веществ cp > cV , а для идеального газа справедливо:
cp = cV + R
Для данного процесса справедливо выражение:
5. Адиабатическое расширение
Данный процесс осуществляется без подвода теплоты, если первоначально газ сжат и находится в термически изолированном сосуде. Здесь газ расширяется и охлаждается при условии p1>p2.
В этом процессе работа осуществляется за счет убыли внутренней энергии газа.
Q = 0
dU = δQ — δW
dU = -δW
∆U = -W
Из показанного ранее выражения cV = dU/dT следует, что dU = cVdT. И получаем:
Полученное выражение справедливо для 1 моль вещества. Для несколько молей, получаем:
Выражение для работы расширения при адиабатическом процессе:
Для данного процесса справедливы выражения:
Формулы, выведенные для всех изопроцессов, справедливы и для обратных процессов.


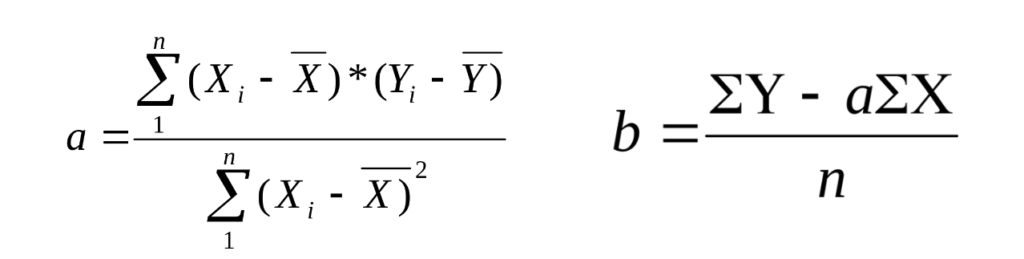
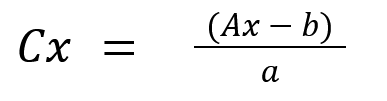

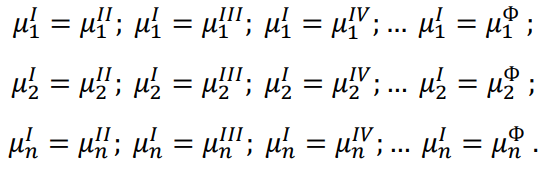








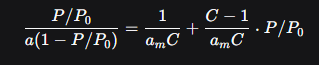
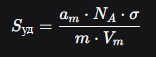

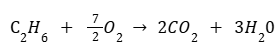
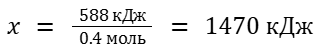
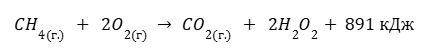
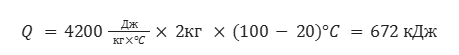
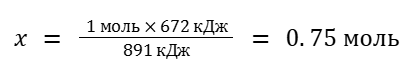












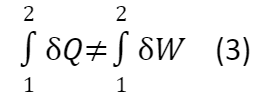
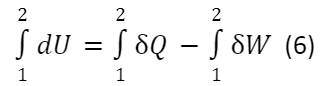

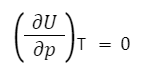
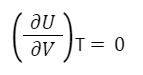

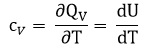
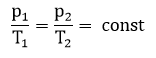
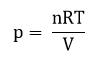
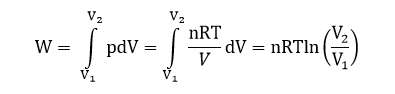
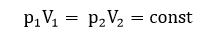

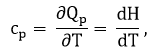

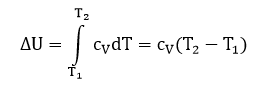
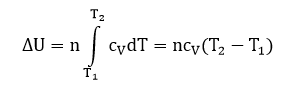
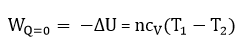
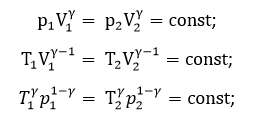
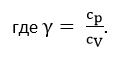
Новости
22.11.2024
22.11.2024
24.10.2024
14.10.2024
09.10.2024
01.10.2024